Reference: April 2024 | Issue 4 | Vol 10 | Page 28
Interstitial lung disease (ILD) comprises a diverse range of lung conditions characterised by inflammation and fibrosis of the lung interstitium. A significant proportion of patients diagnosed with ILD have pulmonary fibrosis (PF), with idiopathic pulmonary fibrosis (IPF) recognised as the most common type, constituting 17-to-37 per cent of all ILDs. A proportion of ILD patients may develop a progressive fibrotic phenotype, which may collectively be called progressive pulmonary fibrosis (PPF), previously known as progressive fibrosing ILD (PF-ILD). PPF represents an ILD with radiological signs of fibrosis and shows evidence of progression over time (Figure 1).
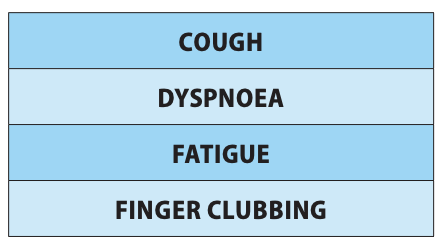
Due to the progressive nature of IPF/PPF, patients frequently experience a range of debilitating symptoms which not only vary in severity, but also worsen over time. These symptoms include worsening breathlessness, persistent cough, fatigue, and adverse effects on emotional and psychological wellbeing.
The disease burden in IPF/PPF manifests through diverse impacts on physical, social, and emotional wellbeing, presenting formidable challenges for individuals and healthcare systems. Patients living with IPF/PPF experience an unpredictable disease trajectory and substantial symptom burden which collectively impose a profound impact on both their own and their caregivers’ quality-of-life.
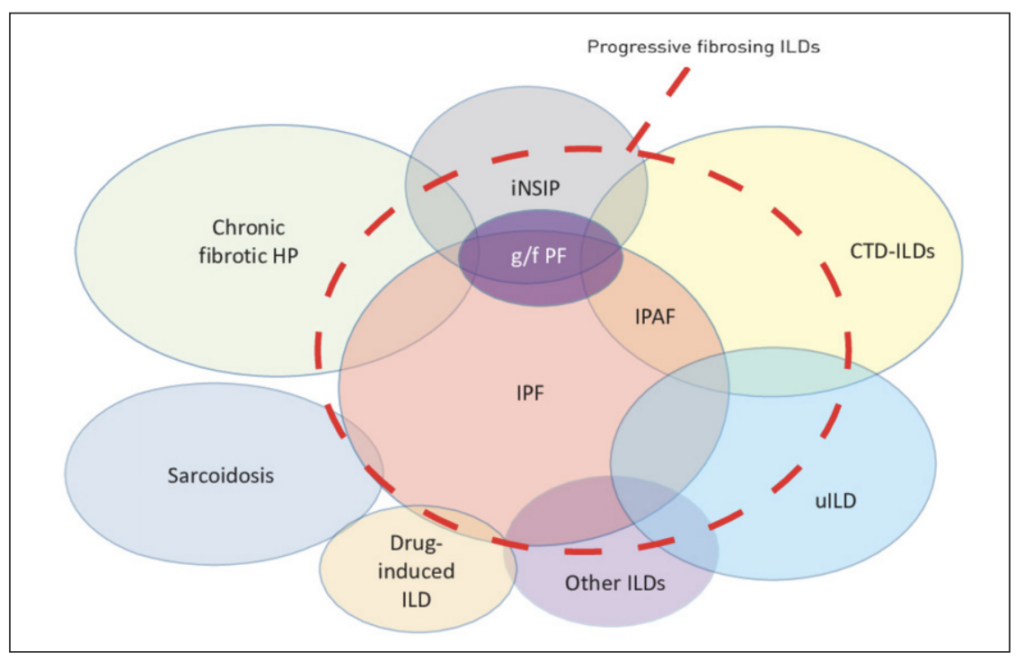
FIGURE 1: Schematic representation of types of ILD that may be associated with a progressive fibrosing phenotype. Connective tissue disease (CTD)-ILDs include rheumatoid arthritis-associated ILD, systemic sclerosis-associated ILD, mixed CTD-associated ILD, and other autoimmune ILDs. Other ILDs include exposure-related ILDs (asbestosis and silicosis), non-idiopathic pulmonary fibrosis, idiopathic interstitial pneumonias (desquamative interstitial pneumonia, etc), and others. (Abbreviations: G/f PF: Genetic and/ or familial pulmonary fibrosis; HP: Hypersensitivity pneumonitis; iNSIP: Idiopathic nonspecific interstitial pneumonia; IPAF: Interstitial pneumonia with autoimmune features; uILD: Unclassifiable ILD Reproduced with permission of the ©ERS 2024: European Respiratory Review 28 (153) 190109; DOI: 10.1183/16000617.0109-2019. Published 1 October 2019
Epidemiology
The incidence of IPF notably increases with advancing age, with higher prevalence rates observed in males. However, there exists considerable international heterogeneity concerning the incidence and prevalence of IPF, partially attributable to inconsistencies in diagnostic coding practices, resulting in a notable gap in comprehensive epidemiological reporting.
Estimates suggest that the prevalence of IPF, the most common ILD, ranges widely throughout Europe and North America from 1.3-to-42.7/100,000 persons, and an estimated incidence of 2.8-to-19.0/100,000 person-years (Table 1). In Ireland it is estimated that 1,000 patients are affected by IPF, although it is widely regarded that this is a conservative estimate and that the prevalence of IPF is much higher. The median survival from diagnosis is 4.5 years.
Estimated prevalence of non-IPF PPF ranges from 6.9 (Europe) to 70.3 (US)/100,000 persons, and the estimated incidence ranges from 2.1 (Europe) to 32.6 (US)/100,000 person-years.
High symptom burden
IPF/PPF is an unpredictable disease with a variable course, impacting both physical and psychological wellbeing. Patients diagnosed with IPF commonly experience symptoms such as worsening breathlessness, cough, and fatigue, significantly affecting their health-related quality-of-life (Figure 2). These symptoms can induce distress and impose an overwhelming burden to both patients and caregivers.
Patients with IPF may experience a range of comorbidities, further adding to the burden of the condition, and may incorporate pulmonary or extrapulmonary comorbidities including coronary heart disease, diabetes mellitus, pulmonary hypertension, chronic obstructive pulmonary disease (COPD), obstructive sleep apnoea, gastroesophageal reflux, and lung cancer. Moreover, side-effects from medication, particularly antifibrotic treatment, can further exacerbate the disease burden and impact quality-of-life.
The diverse array of needs faced by patients with IPF/PPF requires a comprehensive multidisciplinary approach to care, encompassing equal access to supportive care, routine monitoring, and the need for repeated hospitalisation – particularly at the end-of-life stages. Integrated services that prioritise safety, timeliness, and patient-centredness are essential.
The urgent imperative for early and accurate diagnosis and referral to specialist centres cannot be overstated. National initiatives such as the Patient Charter for IPF developed in collaboration with the Irish Lung Fibrosis Association (ILFA), and the IPF Position Statement by the Irish Thoracic Society, underscore the necessity for rapid referral to centres with expertise in IPF/PPF. Delays in diagnosis and referral may result in avoidable negative impacts for patients, including delayed access to pharmacological interventions and supportive treatments. Alarmingly, reports indicate that referrals to specialised services can occur more than 12 months post-initial diagnosis, highlighting a significant issue given the progressive nature of IPF/PPF.
| IPF | NON-IPF PPF | ||
|---|---|---|---|
| EUROPE/US | Estimated Incidence | 2.8-to-19.0/100,000 person-years | 2.1 (Europe) to 32.6 (US)/100,000 person-years |
| Estimated Prevalence | 1.3-to-42.7/100,000 persons | Range from 6.9 (Europe) to 70.3 (US)/100,000 persons |
TABLE 1: Estimated incidence and prevalence of IPF/non-IPF PPF
Psychological support
There is a glaring deficit in the provision of psychological support for patients diagnosed with IPF/PPF, with reports of fragmented access to such services nationally. The psychological impacts of these conditions are wide-ranging and encompass evidence that patients may experience anxiety and depression at various stages in the disease trajectory. In one study, prevalence rates of depression and anxiety were very high in patients with ILD, reaching 49 per cent and 60 per cent, respectively.
Despite these numbers, the rates of onward referral to a clinical psychologist remained low, reflecting the reality faced by many patients with IPF/PPF. Another study reported that a mere 6 per cent of patients diagnosed with pulmonary fibrosis were referred to a clinical psychologist, with all referrals being made by the patients’ consultants occurring more than 12 months after diagnosis. The recognition of psychological needs and the subsequent referral to appropriate clinical services remain underutilised, highlighting an essential role for healthcare professionals in facilitating referrals to mental health supports.
Information needs
Internationally, there remains a significant gap in meeting information needs for both patients with IPF/PPF and their caregivers. Recognising patients as partners in care necessitates the timely delivery of clinically appropriate information covering a diverse range of topics. Patients and their families require bespoke timely delivery of information on medication use and potential side-effects, supplemental oxygen use, nutrition and exercise, management of symptoms, travel advice (particularly for those on oxygen), trusted online information resources, legal advice, and end-of-life planning.
Timely delivery of clinically appropriate information is a cornerstone in the optimal management of patients with IPF/PPF and could be optimised through the implementation of a national clinical care programme, which would support the provision of comprehensive information with specific attention to the content, structure, format, and timing of information delivery.
Access to services
It appears that there are significant challenges in providing integrated clinical care for individuals with IPF/PPF. International best practice dictates that integrated clinical care programmes for people with chronic conditions optimise quality healthcare, incorporating the full spectrum of care and services that are coordinated across different providers and healthcare sites, and which significantly place people at the centre of care.
Our recent scoping review confirmed key areas of concern including access to accurate and timely diagnosis, referral for lung transplantation assessment, access to pulmonary rehabilitation, pulmonary function testing, access to allied health professionals, and referral to palliative care.
The absence of a national clinical care programme for patients with IPF/PPF exacerbates the fragmentation of care. The lack of a cohesive approach increases the likelihood of negative impacts on patients and raises concerns about the possibility of individuals falling through the gaps in the healthcare system. Without a unified programme, patients may face challenges in receiving early and accurate diagnosis, as well as accessing essential healthcare staff and support services.
Cassidy and colleagues (2022) highlight the poor access to specialist and multidisciplinary healthcare for patients with PF nationally. This deficiency underscores the need for a comprehensive and coordinated approach to care that addresses the complex needs of patients with IPF/PPF.
Furthermore, there can be inequitable access to services across geographical locations, evidenced in a significant gap between optimal and actual access to care. Recent evidence suggests that access for patients with current PF to specialist nursing care was impacted by geographical location, with a broad variation seen nationally, ranging from 37-to-80 per cent, with several locations not having access to a nurse specialist. A national clinical care programme could play a crucial role in standardising care practices, improving access to specialised services, and ensuring continuity of care across different healthcare settings.
Technology advancements
Recognising the role of technology in delivering high quality healthcare is increasingly important, especially in the context of managing chronic conditions like IPF/PPF. This was further highlighted during the Covid-19 pandemic. Integrating technology into clinical care programmes for patients with IPF/PPF can significantly enhance the delivery of care and improve patient outcomes. Several key aspects of technological integration into IPF/PPF
care programmes include:
1. Remote monitoring of patients’ health status including spirometry, physical activity, symptoms monitoring, assessment of quality-of-life, and medication tolerability.
2. Virtual consultations facilitated by platforms such as ‘Attend Anywhere’ provide opportunities for healthcare providers to connect with patients and other healthcare professionals remotely.
3. Enhanced communication and collaboration: Technology facilitates seamless communication and collaboration among members of the healthcare team, including specialists, GPs, allied health professionals, and patients.
4. Data-driven decision-making: Technology enables the collection and analysis of real-time patient data, empowering healthcare providers to make informed decisions about treatment strategies, monitor disease progression, and identify trends or patterns that may require attention.
By embedding technology into clinical care programmes for patients with IPF/PPF, healthcare systems can leverage innovation to overcome geographical barriers, improve access to specialised care, enhance patient engagement, and ultimately optimise health outcomes. Embracing technological advancements is essential for building resilient and patient-centred healthcare systems capable of delivering world-class care, even in the face of challenges such as the Covid-19 pandemic.
Multidisciplinary approach to the management of patients with IPF/PPF
The Irish Thoracic Society Position Statement on the Management of IPF emphasises the need for specialised expertise and multidisciplinary care of patients with IPF. Both international and national guidelines underscore the importance of multidisciplinary discussion involving expert respiratory physicians, radiologists, and pathologists, as the gold standard for IPF diagnosis and management. Multidisciplinary teams for IPF/PPF care should ideally include a range of healthcare professionals to address the diverse needs of patients.
These may include:
1. ILD nurse specialist: Provides specialised nursing care, education, and support for patients with ILD.
2. Physiotherapist: Assist in optimising lung function, managing breathlessness, and promoting physical activity and exercise tolerance.
3. Palliative care clinician: Offers symptom management, supportive care, and end-of-life care for patients with advanced IPF/PPF, and those experiencing significant symptom burden.
4. Medical social worker: Provides assistance with navigating healthcare systems, accessing resources, and addressing psychosocial concerns.
5. Dietician: Offers dietary advice and support to optimise nutrition and manage potential comorbidities
associated with IPF/PPF.
6. Clinical psychologist: Provides psychological support, counselling, and interventions to address emotional distress, anxiety, depression, and coping strategies.
Despite the recognised importance of multidisciplinary care, evidence suggests that there may be deficits in the appropriate staffing of multidisciplinary teams. A recent national quantitative study reported that 45 per cent of patients with IPF had never received a referral to a specified healthcare service such as physiotherapy, palliative care, clinical psychology, or dietetics.
This highlights a significant gap in care delivery and underscores the need for improvements in access to and integration of multidisciplinary services for patients with IPF/PPF and in particular the provision of ILD nurse specialists as critical to the care of patients with ILD.
Patients with IPF/PPF have limited access to pulmonary rehabilitation courses in the community, resulting in many
patients becoming more deconditioned and developing worsening symptoms and potential lack of fitness for lung transplant assessment.
By prioritising multidisciplinary care and addressing staffing deficits, healthcare systems can better meet the complex needs of patients with IPF/PPF and improve overall outcomes and quality-of-life.
A systems approach to care
A systems approach to care is increasingly recognised as essential in healthcare delivery, particularly in addressing the needs of patients with complex chronic conditions such as IPF/PPF. This approach acknowledges the evolving landscape of healthcare, which emphasises broader participation of community-based services alongside hospital-based care initiatives.
National clinical care programmes, and initiatives like Sláintecare, which promotes community-based care, focus attention on the importance of integrating various healthcare services to provide comprehensive and coordinated care to patients. Patients with IPF/PPF face unique challenges due to the unpredictable nature of the disease course and the lack of a national clinical care programme. The present sole focus on airway disease, COPD, and asthma, for clinical care programmes in respiratory disease is having a severe impact on the quality-of-care for patients living with IPF/PPF, representing a major gap in care provision.
Addressing these unmet needs requires the establishment of a clinical care programme specifically tailored to the needs of patients with IPF/PPF. Such a programme would integrate the core principles of integrated care, aiming to optimise care that is safe, timely, efficient, and convenient for patients. By embedding these principles into the care delivery model, the programme can address some of the geographical barriers patients currently face, ensuring equitable access to high-quality care regardless of location.
A comprehensive clinical care programme for IPF/PPF would involve collaboration among various healthcare providers, including respiratory physicians, nurses, allied health professionals, and community-based services. It would prioritise early diagnosis, multidisciplinary management, access to specialised treatments and services, patient education and support, as well as ongoing monitoring and follow-up.
By adopting a systems approach to care and implementing an integrated clinical care programme for IPF/PPF, healthcare systems can improve outcomes, enhance patient experience, and mitigate the burden of this debilitating condition on individuals and their families.
National registry
Establishing a viable and adequately funded national patient registry for IPF/PPF is crucial for capturing accurate epidemiological information on patients diagnosed with these conditions. Current evidence suggests that there is an underestimation of the true prevalence of IPF/PPF, highlighting the need for more comprehensive data collection. The Irish Thoracic Society previously established a national ILD registry, but the registry could not be sustained due to lack of support and resources. A national registry could incorporate several important purposes:
1. Accurate epidemiological data: Registry data would provide more accurate incidence and prevalence data for IPF/PPF, helping to better understand the scope of the disease burden within the population. This information is essential for informing healthcare planning, resource allocation, and policy development.
2. Identification of trends and patterns: By systematically collecting data on patient demographics, disease characteristics, treatment modalities, and outcomes, the registry can help identify trends and patterns in disease progression and management. This information can guide research efforts, clinical practice guidelines, and quality improvement initiatives.
3. Pharmacological use and pharmacovigilance: Monitoring of pharmacological use and pharmacovigilance as new treatments for IPF/PPF evolve.
4. Support for research and clinical trials: Registry data can serve as a valuable resource for researchers conducting epidemiological studies, clinical trials, and observational research in the field of IPF/PPF. Access to comprehensive, real-world data can facilitate the development of new therapies, treatment strategies, and diagnostic tools.
5. Enhanced patient care: A national patient registry can facilitate communication and collaboration among healthcare providers, allowing for more personalised and coordinated care for patients. Clinicians can use registry data to track patients’ disease trajectories, monitor treatment responses, and identify opportunities for intervention. To ensure the success of the registry, it is essential to secure appropriate funding and resources for its establishment and maintenance.
Additionally, efforts should be made to engage stakeholders, including patients, healthcare providers, researchers, and policymakers in the development and implementation of the registry. Data privacy and security measures should also be carefully considered to protect patient confidentiality and to comply with regulatory requirements. Overall, a national patient registry for IPF/PPF holds great promise for advancing our understanding of these conditions, improving patient care, and, ultimately, reducing the burden of disease on individuals and society.
Conclusion
In conclusion, the evolving landscape of IPF/PPF demands a comprehensive and patient-centred approach to care. Rigorous scientific research, advancements in therapeutics, inter-disciplinary collaborations, and integration of patient perspectives have contributed to a deeper understanding of IPF/PPF and its impacts.
The recognition of the diverse range of needs that patients with IPF/PPF have points to the importance of targeted research efforts to address the unmet needs of this under-represented patient population. Access to care remains a significant concern, with unequal access leading to fragmented and suboptimal care – a trend which is particularly influenced by geographical factors.
Our recent review and ongoing national mixed methods study have shed light on the scope of unmet needs of patients with IPF. These findings inform the need for a nuanced and personalised approach to care that addresses the multi-faceted challenges faced by individuals living with these conditions. Moving forward, it is essential to prioritise equal access to care, tailored support services, and interventions aligned with patient preferences, all of which could improve outcomes and quality-of-life for individuals living with IPF/PPF.
References on request