






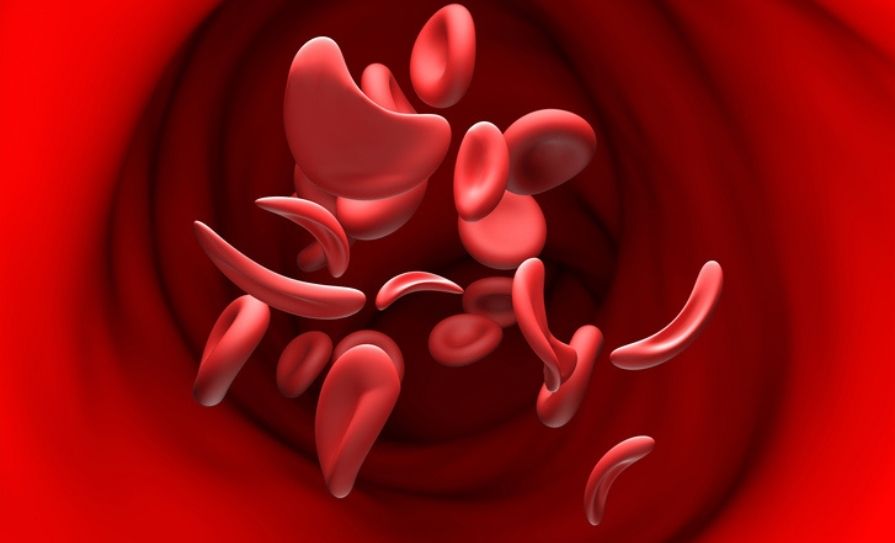
Five studies presented at the 67th American Society of Hematology (ASH) Annual Meeting and Exposition reveal new opportunities to improve care for people living with sickle cell disease, the most common inherited blood disorder.
“These studies allow us to be more accountable to our patients in terms of care and pain management, and to give context about the next phase of sickle cell therapies,” said Prof Titilope Fasipe, Associate Professor of Paediatrics, Baylor College of Medicine, and co-director of the sickle cell and thalassaemia programme, Texas Children’s Hospital, US, who moderated the press briefing ‘Sickle cell disease: From discovery to access’.
“In studying the good, the bad, and the ugly for both our tried-and-true therapies and emerging treatments, we can get the information that our community needs to better inform shared decision-making,” she added.
Several of the studies draw upon real-world data to understand some of the barriers to care access and how various practical factors influence clinical decision making and patient outcomes.
“In our current era, innovation has been defined by cutting-edge advances like gene therapy. However, clinically-meaningful innovation also occurs when we learn new ways to care for patients with the treatments that have been available, like insights on what it means to use hydroxyurea during pregnancy,” said Dr Fasipe.
The first study presents striking evidence of gaps in the delivery of evidence-based care, showing that two-thirds of people visiting US emergency departments for sickle cell pain did not receive timely pain relief in accordance with medical guidelines.
The second study calls for a pragmatic approach to continuing or stopping hydroxyurea for pregnant women, based on findings that taking the drug during pregnancy does not appear to cause specific harms to a developing foetus.
In the third study, researchers analysed long-term outcomes in over 1,000 patients who underwent bone marrow transplant for sickle cell disease, revealing an overall positive outlook for most patients and the best outcomes among younger patients who had a matched related donor.
The final two studies examine gene therapies as a curative option for both sickle cell disease and beta thalassaemia.
The fourth study tracks the clinical practices and timelines involved in implementation of two gene therapies, while the fifth study reports preliminary results from the first clinical trial to examine the gene therapy exagamglogene autotemcel (exa-cel) in children younger than 12 years.
Preliminary results from studies on exa-cel suggest the therapy offers an effective cure for beta-thalassaemia and sickle cell disease in children under 12. Researchers say the therapy’s potential to offer a cure at an early age – before organ damage accumulates – could make exa-cel even more beneficial in children than adults.









Leave a Reply
You must be logged in to post a comment.