







A review of the evolving therapeutic landscape of interstitial lung diseases, exploring the expanding role of precision medicine, multidisciplinary care, and artificial intelligence
Interstitial lung diseases (ILDs) represent a diverse group of lung disorders characterised by varying degrees of inflammation and fibrosis of the lung interstitium. The delicate tissue surrounding the lung’s alveoli becomes progressively fibrosed through prolonged repetitive injury, inflammation, and collagen deposition. This prevents the normal architecture of the lungs from performing gas exchange as intended, reducing oxygen delivery to vital organs and resulting in the onset of progressive, debilitating symptoms of breathlessness and cough. Over time, the lungs become less elastic as fibrosis continues, leading to reduced oxygen availability to tissues and ultimately progressing to respiratory failure.
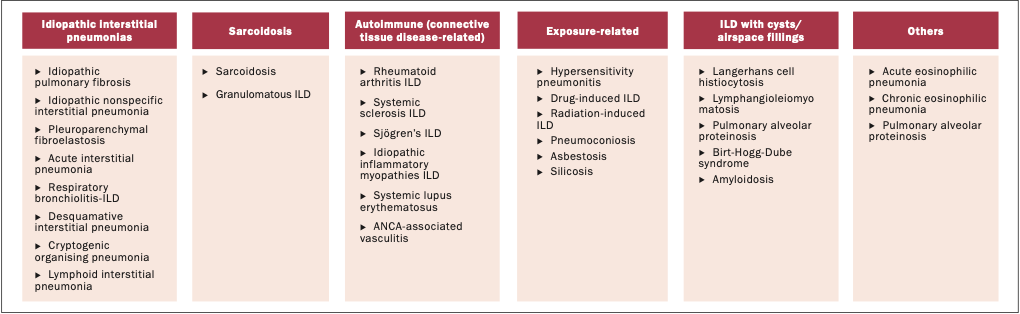
ILDs consist of idiopathic interstitial pneumonias with the commonest being idiopathic pulmonary fibrosis (IPF), ILDs associated with connective tissue diseases (CTD-ILD), which are autoimmune-modulated, ILDs triggered by environmental and occupational exposures (for example, bird and mould exposure and asbestos), drug and radiation-induced ILDs, and sarcoidosis (Figure 1). IPF and progressive fibrotic non-IPF ILDs, termed progressive pulmonary fibrosis (PPF), are associated with the highest morbidity and mortality. Our understanding of the pathophysiology of disease and disease burden has evolved, and with it have come newer approaches to diagnosis and management of ILD.
At present, the current evidence-based treatment focuses primarily on antifibrotic therapy, with pirfenidone and nintedanib forming the standard of care for IPF and selected cases of PPF. However, these therapies slow down, but do not halt, disease progression and are often limited by tolerability issues. This clinical update reviews the evolving therapeutic landscape of ILDs, exploring the expanding role of precision medicine, multidisciplinary care, and artificial intelligence in advancing both diagnostic accuracy and individualised treatment strategies.
Selection criteria
As part of our review, we searched on Cochrane Library, MEDLINE and EMBASE. Terminology searched included: ILD, IPF, CTD-ILD, epidemiology, diagnosis, management, precision medicine in ILD, and multidisciplinary team. Inclusion criteria included randomised controlled trials, systematic reviews, meta-analyses, and guidelines. As this is not a systematic review, the selection of references was at the discretion of the authors.
Incidence and prevalence
ILD encompasses a broad group of chronic restrictive lung diseases and therefore the accurate evaluation of the epidemiology of ILDs is compounded by the varying diagnostic criteria utilised, methodological differences, and differing populations explored between studies. A review of 17 studies with varying methodologies reported wide differences in the measured incidence and prevalence of ILDs. Across these studies, incidence rates ranged from one to 31.5 per 100,000 person-years, while prevalence estimates varied from 6.3 to 71 per 100,000 individuals. In North America and Europe, IPF and sarcoidosis emerged as the most common ILDs. In contrast, hypersensitivity pneumonitis was relatively more frequent in parts of Asia, particularly in India (10.7–47.3%) and Pakistan (12.6%). Amongst the many types, IPF, CTD-ILD, and sarcoidosis are the most common.
A global analysis estimated the burden of sarcoidosis to be 43.07 million prevalent cases and 23.2 million incident cases worldwide. Country-specific data for IPF suggests a prevalence of 6.8 million in the US and 4.6 to 7.4 cases per 100,000 people per year across the European Union. A recent review on the global epidemiology of ILD provides the most up-to-date review of the literature, highlighting the regional differences that exist when estimating incidence and prevalence of different subtypes of ILD. The authors highlight the varied methodological approaches utilised to estimate the incidence and prevalence of ILDs, ranging from claims databases to registry reports, and how this leads to a heterogeneity in findings. Despite these limitations, there is an appreciation from most studies that there is an overall increase in the incidence and prevalence of fibrotic ILDs over time.
This comprehensive review found US incidence rates of IPF to be higher than European studies, ranging from 7–17 per 100,000 versus 0.2–3 per 100,000 population in Europe. The incidence range for sarcoidosis varied according to geography and methodology, ranging from 0.13 to 17.8 per 100,000 and for hypersensitivity pneumonitis, the incidence was lowest at 0.3–0.9 per 100,000 cases.
In comparison to other respiratory illnesses such as asthma and chronic obstructive pulmonary disease (COPD), ILDs represent a significantly lower global disease burden. Studies indicate that 55 per cent of ILD patients initially receive an incorrect diagnosis, with an additional 38 per cent being misdiagnosed at least twice. This misdiagnosis contributes to delayed treatment and the potential for irreversible disease progression. Therefore, although ILD is less common than other chronic respiratory diseases, early and accurate diagnosis is crucial. Prompt recognition can substantially reduce permanent damage to lung architecture, thereby preserving normal gas exchange and improving long-term outcomes.
Diagnosis
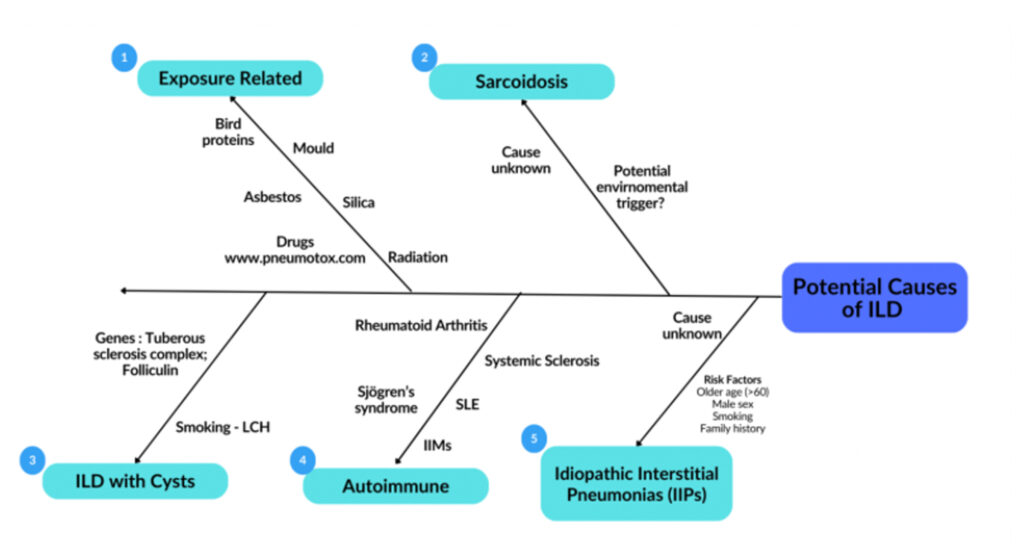
A critical aspect of diagnosing an ILD begins with a thorough clinical history and physical examination. Patients typically report progressive shortness of breath, which is initially noticeable on exertion and a chronic non-productive cough. Given the wide differential and heterogeneity in subtypes of ILDs, it is important to take a thorough history of potential aetiologies for the underlying ILD including exposures, occupations, medication history, family history of ILDs and features of CTD (Figure 2). Physical examination typically reveals finger clubbing and fine inspiratory crackles on auscultation of the lungs. Additional clinical signs may be observed depending on the underlying aetiology. Examples include signs indicative of an underlying CTD such as sclerodactyly, telangiectasia, skin puckering around the mouth and a photosensitivity rash. Similarly, skin thickening may be observed in systemic sclerosis, as well as rheumatoid nodules in rheumatoid arthritis (RA). An in-depth history and examination provide vital information to diagnose and subsequently subtype the ILD. This then aids management and provides valuable information on prognosis. It is for this reason that the power of history and examination should not be underestimated and should be completed to a high standard. Due to the non-specific nature of early symptoms and physical examination findings, diagnosis is often delayed by several months, with the average case being diagnosed after 12 months. As previously discussed, conditions such as COPD are frequently considered, or even misdiagnosed, prior to an accurate identification of an ILD.
Autoimmune serology is an important diagnostic investigation in ILDs in the presence or absence of coexistent symptoms that may suggest an underlying CTD. An ILD can be an initial manifestation of an underlying CTD. Approximately 15 per cent of new diagnoses of ILDs are later diagnosed as having a CTD. Positive autoimmune serology in ILD warrants a rheumatological assessment to evaluate the presence of a previously undiagnosed CTD, as a shift in diagnosis from an idiopathic subtype of an ILD to a CTD-ILD has a major impact on therapeutic strategies and prognosis. However, positive autoimmune serology alone is not sufficient to diagnose a CTD and thus this requires robust rheumatological assessment and expertise with knowledge of the established diagnostic classifications for the varied CTDs. Positive autoimmune serology in ILD without symptoms or signs indicative of a CTD has been given the research terminology of interstitial pneumonia with autoimmune features (IPAF). This diagnostic label has a number of challenges for the patient, as there is limited evidence regarding monitoring or therapeutic strategies in this cohort of patients.
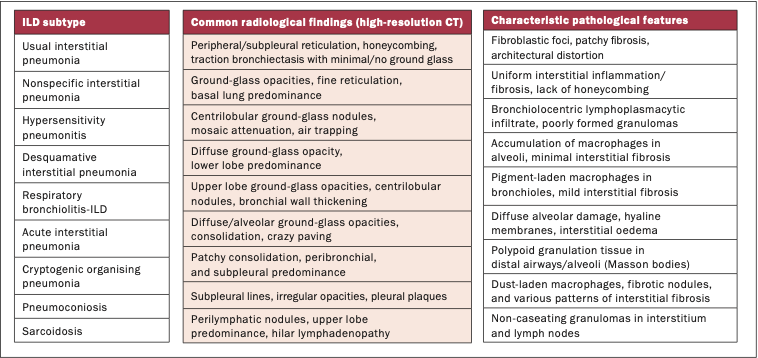
High-resolution CT (HRCT) is pivotal for the diagnosis of ILD. Within the last decade, there has been an increasing appreciation that ILDs encompass a broad spectrum of diseases with distinct radiopathological features (Figure 3). This has facilitated the diagnosis of ILDs to become more accurate and standardised. IPF has a typical pattern on HRCT known as the usual interstitial pneumonia (UIP) pattern. It is defined as a subpleural basal predominant ILD characterised by reticulation and honeycombing with minimal or no ground glass. Although this pattern is typical for IPF, it is also the most common pattern seen in RA-associated ILD (RA-ILD). Whereas non-specific interstitial pneumonia (NSIP) pattern, characterised by basal predominant ground glass and reticulation, is more commonly observed in CTD-ILDs such as systemic sclerosis (SSC)- or myositis-associated-ILDs. Recognition and diagnosis of these diverse patterns can be challenging to assess, even for expert radiologists and clinicians. Thus, a multidisciplinary approach to diagnosing ILD is essential.
The role of the multidisciplinary team (MDT)
The heterogeneous clinical, radiological, and pathological features of ILD make it exceptionally challenging for diagnosis, and therefore, an MDT approach has been employed as a gold standard and advocated for ILD diagnosis. International guidelines recommend MDT discussion as the reference standard for ILD diagnosis, reflecting the need to integrate expertise from pulmonology, radiology, and pathology. A multicentre evaluation comparing individual clinician diagnosis to an MDT approach to diagnosis found that an MDT approach significantly improved the diagnostic accuracy. In this study, seven expert MDTs across Europe first evaluated 70 ILD cases independently without consultation, giving differential diagnoses and likelihoods of diagnosis, and then convened in an MDT to discuss the cases again. Inter-MDT agreement for overall diagnosis was higher than inter-observer agreement, particularly for IPF. MDTs made the diagnosis with more confidence than individual clinicians or radiologists alone. Furthermore, in a study of 404 physicians from 57 countries independently assessing 60 ILD cases without access to MDT consultation, and their diagnostic performance being compared with that of 34 international ILD experts. Diagnostic agreement for IPF was significantly higher among experts (weighted κ=0.65, IQR 0.53–0.72) than among academic physicians (κ=0.56, IQR 0.45–0.65) or physicians who reported access to MDT meetings (κ=0.54, IQR 0.45–0.64). Crucially, when prognostic accuracy was considered, experienced non-university physicians who regularly attended MDT meetings had similar accuracy to that of experienced academic physicians.
These findings highlight two key roles of the MDT discussion. First, MDTs reduce variability in diagnostic decision-making, ensuring that non-specialist or community physicians achieve a diagnostic accuracy on par with international experts. Second, MDT involvement directly impacts patient outcomes by improving prognostic discrimination, thereby guiding appropriate treatment strategies such as antifibrotic therapy and clinical trial enrolment. Without MDT support, diagnostic performance is less reliable, which may contribute to misclassification and treatment delays. Taken together, this evidence demonstrates that MDT discussion is not only a best-practice recommendation, but a practical necessity for high-quality ILD care. Expanding access to MDT infrastructure, particularly outside of specialist or academic centres, is essential for reducing disparities in diagnosis and management worldwide. A Delphi study of over 130 ILD experts highlighted important desirable aspects of an MDT, including membership, environment, and technology and reporting of the MDT. A number of expert recommendations were made and defined as essential, highly desirable, and desirable features of an MDT. Some of these included essential components such as a quiet environment with good quality audio-visual equipment, reviewing high-quality HRCT with at least one radiologist present in the MDT, and the use of standardised templates to document discussions. Highly desirable aspects include membership with at least one pulmonologist and pathologist, with a member with at least five years’ experience or training in ILD.
The role of the MDT is therefore paramount, as we cannot rely on imaging alone. MDT diagnosis encompasses a discussion of the patient’s history, radiological findings, immunology, and sometimes pathology. Guidelines now more clearly define the diagnostic pathway for ILDs and the MDT approach is considered gold standard.
Precision medicine
Precision medicine uses an individualised approach to treating patients. It considers each patient’s specific clinical characteristics and findings in order to deliver precise and targeted treatment. It has been described as an ‘individualised multidimensional assessment for distinct traits that can be targeted by specific interventions’. Although clinical trials provide evidence on the efficacy and side-effects of medications, these statistics are based on population effects and not individual responses. In reality, there is a wide range of individual responses when it comes to how effective a drug is and the side-effects experienced. Targeting the right drug to the right patient can maximise efficacy and survival, while minimising adverse effects and overall costs.
To achieve this, we need a better understanding of which features can predict treatment response. This is currently being investigated through several approaches. Biomarkers play a major role in classifying ILD, predicting prognosis, and monitoring response to treatment. Molecular biomarkers involved in ILD can be divided into three categories based on the underlying mechanism: Epithelial cell dysfunction and senescence, aberrant immunity, and abnormal lung remodelling. For example, the biomarker periostin (linked to abnormal lung remodelling) may be useful in diagnosis, prognosis, and monitoring treatment response. In contrast, HSP70 (associated with aberrant immunity) is useful only for prognosis. There are increasing numbers of exploratory studies evaluating the potential of varied molecular biomarkers in the diagnosis and prognostication of ILD. However, to date, none of these individual biomarkers has been validated for clinical practice. KL-6, an epithelial biomarker, is the most widely studied biomarker and is used clinically in the diagnostic pathways in countries such as China. It has not been adopted in US or European guidelines due to a lack of robust validated evidence of utility.
Novel radiological biomarkers used to improve diagnosis and predict disease progression are of particular interest. HRCT is already a crucial tool in the assessment of ILD, but high levels of interobserver variability make it difficult to produce consistent and reliable results. Deep learning (DL), a widely used form of artificial intelligence (AI) and machine learning (ML), allows for the analysis of large-scale and high dimensional data sets. It provides strong image recognition capabilities through pattern matching which can be utilised in the assessment of medical scans. A systematic review evaluated the effectiveness of using DL-based AI to analyse ILD on HRCT. The authors conducted a comprehensive literature search that included all studies that applied DL to ILD analysis on chest CT. It was concluded that DL has demonstrated potential for diagnosing and assessing ILD. DL achieved an accuracy of 78–91 per cent with the two highest performing studies reaching near expert level performance. Although these results are promising, many studies exhibited a high risk of bias due to concerns around study design and data selection. Therefore, the evidence is not yet sufficient for widespread clinical use, highlighting that there is certainly potential for the use of AI in assessing ILDs in the future. However, there remains a need for further development.
Furthermore, studies have looked at the utility of early CT findings that could predict future disease progression in ILD. Recent innovations in computer analysis of CT imaging have been made possible by rapid advances in computer technology. Novel algorithms now allow for precise quantification of parenchymal patterns using three-dimensional volumetric CT datasets. These measures have proven to more accurately predict patient survival in various fibrosing lung diseases in comparison to traditional visual assessment of CTs. In addition, computer-based analysis has also been developed to quantify vessel-related structures (VRSs). VRSs include pulmonary vasculature and its associated structures, such as perivascular fibrosis. This quantification has been strongly linked with survival prediction in patients with IPF. This represents a significant advancement in the use of AI for disease diagnosis and prognosis in ILD, particularly in detecting subtle patterns not visible by the human eye.
In ILD, precision medicine involves assessing genetic and molecular profiles, diagnosis, staging, prognosis, and targeted pharmacotherapies. The treatable traits model is often used in this context. This model is based on three core criteria: The trait must be clinically important (associated with adverse outcomes), recognisable and measurable (using tools such as phenotyping), and treatable. In ILD, the treatable traits assessed include disease aetiology, pulmonary and extrapulmonary manifestations, and lifestyle factors. Overall, precision medicine plays a key role in the treatment, prognosis, and monitoring of disease progression in ILD.
Current treatments
The management of ILD aims to target the underlying pathogenic process. Due to the varied inflammatory and fibrotic components of ILD, current therapies are designed to either reduce inflammation or target fibrosis, resulting in distinct therapeutic outcomes. Another approach in the treatment of ILD is the use of immunosuppressants. Despite a lack of robust clinical trial evidence, immunosuppressants are reserved for ILDs with a predominantly inflammatory driver and phenotype. These include hypersensitivity pneumonitis, cellular nonspecific interstitial pneumonias (NSIP) and CTD-ILD. On the other hand, antifibrotics have a robust evidence-base in the treatment of IPF and PPF. It is essential to complement these therapies with non-pharmacological strategies that aim to reduce the severity or alleviate breathlessness.
Dyspnoea is an extremely common symptom experienced by patients with ILD, with studies showing that 68.2–98 per cent of patients experience it. The primary non-pharmacological intervention employed for the management of ILD is pulmonary rehabilitation. Pulmonary rehabilitation has been found to improve functional exercise capacity and health-related quality-of-life, both short-term and long-term. A Cochrane review of 12 studies demonstrated that pulmonary rehabilitation improved how far patients could walk in six minutes by 40 metres short-term. The improvement was sustained by patients walking an extra 37 minutes at six to 12 months post pulmonary rehabilitation. Additionally, improvements were also found in improving maximum exercise capacity, reducing dyspnoea and improving health-related quality-of-life. However, the long-term impact of pulmonary rehabilitation in ILD is still not well-supported and is a topic of ongoing research.
Another non-pharmacological measure that has been proven to be beneficial is handheld fan therapy (HHF). A mixed-methods pilot study looked at the quantitative and qualitative results of using fan therapy in the management of ILD. The study involved 30 adults with fibrotic ILD experiencing moderate-to-severe dyspnoea. A series of qualitative interviews with patients revealed that there was initial apprehension and scepticism about the benefit it would provide. However, patients found that there indeed was a symptomatic benefit of using fan therapy. The patients who were on oxygen therapy reported some improvement, but relief was not as effective as their usual oxygen therapy, despite being easier to use. Although some subjective improvement was noted by patients, quantitative data did not show a significant improvement in dyspnoea-12 scores. The conclusion of this study is that HHF was perceived as helpful by patients to manage dyspnoea; however, efficacy remains uncertain, likely due to the short trial duration and small sample size.
Furthermore, ambulatory oxygen has been explored as a measure to improve breathlessness. The AmbOx study is a trial conducted in the UK, which explored the use of ambulatory oxygen in the management of fibrotic ILD. A total of 84 patients with varied fibrotic ILDs, the commonest IPF, who were not hypoxic at rest, but desaturated during a six-minute walk test to 88 per cent or below, were recruited in the study. Patients were allocated to two weeks of ambulatory oxygen or no oxygen therapy. Results were measured by looking at improvement in health-related quality-of-life using the King’s Brief ILD questionnaire (K-BILD). K-BILD score was on average 55.5 ± 13.8 in the ambulatory oxygen group compared to 51.8 ± 13.6 in the no oxygen group, highlighting significant improvement, specifically in the breathlessness and activity and chest symptom sub-domains, but not the psychological subdomain.
Pharmacological therapies are utilised in reducing disease progression through immunosuppression or reducing fibrosis. Much of the role of immunosuppressant use in predominantly inflammatory ILDs has been extrapolated from randomised control trials (RCTs) in SSC-ILD and open-label studies. Immunosuppressive agents, including mycophenolate mofetil and azathioprine, are employed in CTD-ILDs. While evidence from large RCTs is limited, observational studies found modest improvement in lung function, particularly in SSC-ILD and RA-ILD.
The Scleroderma Lung Study I explored the use of cyclophosphamide for one year followed by a year off treatment versus placebo in individuals with SSC-ILD. After 12 months, forced vital capacity (FVC) showed a significant improvement of 2.53 per cent in the cyclophosphamide group compared to the no treatment group. Subsequently, the Scleroderma Lung Study II compared the use of mycophenolate mofetil for two years to cyclophosphamide for one year, followed by a placebo for the second year. This non-inferiority study found consistent efficacy with both mycophenolate and cyclophosphamide, with improvements in FVC of 2.17 per cent and 2.86 per cent, respectively. There were fewer adverse effects and better tolerability with mycophenolate therapy, and thus, based on these studies, mycophenolate is considered first-line therapy in SSC-ILD.
The evidence of the role of immunosuppression in RA-ILD is less robust and not clear-cut. The commonest pattern of ILD in RA-ILD is a UIP pattern, which is also a feature of IPF. In IPF, immunosuppression has been shown to be harmful, as evidenced by increased hospitalisations and death in the interim analysis of the PANTHER study. This concern is supported by the results of the PANTHER-IPF trial, a randomised controlled study that tested the combination of prednisone, azathioprine, and N-acetylcysteine in patients with IPF, a disease characterised by the UIP pattern. An interim analysis demonstrated that patients receiving this regimen had significantly higher rates of death (eight vs one) and hospitalisation (23 vs seven) compared with placebo, without any evidence of improvement in lung function or clinical outcomes. These findings led to early termination of the combination arm, highlighting that broad immunosuppression in UIP may not only lack benefit, but also accelerate harm, likely through increased susceptibility to infection, impaired host repair mechanisms, and treatment-related toxicities. Clinicians are therefore cautious with the use of immunosuppression in RA-UIP without robust evidence of efficacy and lead with caution while considering combination therapies involving immunosuppression.
Pirfenidone (PFD) and nintedanib are two antifibrotic agents licensed for the management of IPF. PFD is an oral, antifibrotic medication that has several mechanisms of action, including reducing oxidative stress, downregulating the TGF-β1 and inhibiting the production and release of inflammatory cytokines. PFD suppresses TGF-β1 and other growth factors, and as a result, downregulates downstream inflammatory pathways. Furthermore, PFD was found to inhibit redox reactions and control oxidative stress-related genes and enzymes. Furthermore, emerging data show the PFD suppresses inflammasome signalling components and DAMPs (danger-associated molecular patterns) and oxygen-free radicals. Therefore, the mechanism of action of PFD is unique in that it has both antifibrotic and immunosuppressive effects through the regulation of immune cells. The CAPACITY trials 004 and 006 and the ASCEND trial’s combined data show that PFD slowed down the decline of lung function, improved progression-free survival, and decreased death rate over a period of 12 months. Adverse events include fatigue, loss of appetite, weight loss, and photosensitivity rash.
Nintedanib is a multi-targeted, intracellular tyrosine kinase inhibitor utilised worldwide for the management of ILD due to its antifibrotic properties. It exerts its therapeutic effects by inhibiting several tyrosine kinases, including platelet-derived growth factor (PDGF) receptors, vascular endothelial growth factor (VEGF) receptors, and fibroblast growth factor (FGF) receptors, thereby disrupting key signalling pathways involved in fibroblast proliferation and extracellular matrix (ECM) deposition. Although the precise mechanism of action remains not entirely understood, studies using fibroblasts isolated from the lungs of patients with IPF have demonstrated that nintedanib downregulates the expression of ECM proteins such as collagen and fibronectin, and inhibits transforming growth factor-beta 1 (TGF-β1)-induced myofibroblast differentiation. Additionally, nintedanib has been shown to induce ATG7-independent autophagy in lung fibroblasts, contributing to its antifibrotic effects. Interestingly, while morphological changes were observed in fibroblasts, the drug did not induce apoptosis.
The INPULSIS trials of nintedanib in IPF – the INPULSIS-1 and INPULSIS-2 phase 3 trials – evaluated the efficacy and safety of nintedanib (150mg twice daily) in 1,066 patients with IPF. The primary endpoint, annual decline in FVC was significantly reduced in both trials. In INPULSIS-1, the adjusted annual rate of FVC decline was −114.7ml with nintedanib versus −239.9ml with placebo (difference 125.3ml; 95% CI, 77.7–172.8; P<0.001). In INPULSIS-2, the respective values were −113.6ml versus −207.3ml (difference 93.7ml; 95% CI, 44.8–142.7; P<0.001). Therefore, these studies show that nintedanib significantly slows the annual decline in lung function, as measured by FVC, in patients with IPF. The INPULSIS trials therefore establish nintedanib as an effective antifibrotic therapy that modifies disease progression in IPF. Its ability to reduce lung-function decline provides a crucial therapeutic option in a condition with limited treatments. Incorporating nintedanib into routine clinical practice can improve long-term patient outcomes, though proactive management of gastrointestinal side-effects is essential to maintain adherence.
Furthermore, the INBUILD trial, a randomised, double-blind, placebo-controlled, parallel-group study conducted across 153 sites in 15 countries, evaluated the efficacy of nintedanib in patients with various progressive fibrosing ILDs, including chronic hypersensitivity pneumonitis, autoimmune-related ILDs, idiopathic non-specific interstitial pneumonia (iNSIP), unclassifiable idiopathic interstitial pneumonia, and other ILDs. The trial demonstrated a consistent benefit of nintedanib in reducing the rate of FVC decline compared with placebo across all subgroups, with mean differences (mL/year) of: Chronic hypersensitivity pneumonitis, 73.1 (95% CI -8.6 to 154.8); autoimmune ILDs, 104.0 (21.1 to 186.9); iNSIP, 141.6 (46.0 to 237.2); unclassifiable ILD, 68.3 (-31.4 to 168.1); and other ILDs, 197.1 (77.6 to 316.7). There was no significant interaction between treatment effect and disease subgroup (p = 0.41). Over the 52-week study period, nintedanib significantly slowed the rate of FVC decline in patients with progressive fibrosing ILDs. Nintedanib is thus licensed for both IPF and PPF, with pirfenidone licensed in IPF alone, and are part of international guidelines.
These findings have prompted further investigation into combination therapy approaches. Shumar et al suggest that the integration of nintedanib with pirfenidone could offer synergistic benefits by addressing both fibrotic and inflammatory components of ILD pathophysiology. This potential hypothesis warrants further investigations in clinical trials with combination therapies; however, that may be compounded by the cumulative costs of both therapies and adverse events.
In summary, a multifaceted approach is essential in managing the complex pathology of ILD. The pharmacological management of ILD requires targeting both inflammatory and fibrotic mechanisms. Immunosuppressants remain important for ILDs with predominantly inflammatory drivers, while antifibrotic agents such as pirfenidone and nintedanib have demonstrated strong evidence for slowing disease progression in IPF and progressive fibrosing ILDs. Supportive strategies, including pulmonary rehabilitation, ambulatory oxygen, and HHF therapy, provide additional benefit, although their long-term impact requires further study. These findings highlight the need for continued innovation, including combination and novel therapies, to address the complex and heterogeneous nature of ILD.
Treatment advances
The clinical trial landscape for ILD is evolving, with a need for safer, more effective therapies in addition to current treatment. Advancements in treatment offer the opportunity for new novel therapies, optimisation of current treatment strategies, and targeted treatment for individuals earlier in the disease cycle. Current treatments do not halt disease progression, and gastrointestinal side-effects often hinder the delivery of these therapies.
Currently, there are over 2,000 studies reported on the clinical trials website, clinicaltrials.gov, spanning early phase through to phase 3 studies. It is particularly exciting to see some of the most promising trials at phase 2a now progress to phase 2b, and whilst it is out of the scope of this review to describe all these studies, we have chosen a few as examples of the emerging therapeutic landscape in IPF and other ILDs.
One of the most promising treatment advances in the last decade is the advent of nerandomilast in patients with IPF and PPF (41–42). Nerandomilast is a preferential inhibitor of phosphodiesterase 4B, administered orally, and which provides antifibrotic and immunomodulatory effects, and was approved by the US FDA in late 2025. A 12-week phase 2a RCT (NCT05321069) with 147 IPF patients showed encouraging results with a median FVC difference of 88.4mL with nerandomilast without antifibrotics (95% CI: 29.5–154.2; 99.8% probability of superiority) and 62.4mL in those on antifibrotics (95% CI: 6.3–125.5; 98.6% probability). This led to the phase 3 FIBRONEER-IPF (NCT05321069) and FIBRONEER-ILD trials (NCT05321082) (41–42). Both studies were double-blind, multicentre RCTs involving 1,176 IPF patients and 1,172 non-IPF progressive fibrosis patients, respectively. Both trials demonstrated that nerandomilast significantly reduced the rate of FVC decline (with no significant differences between those on or off antifibrotics). In the FIBRONEER-IPF study, a nerandomilast/pirfenidone interaction complicated interpretating the efficacy results by possibly altering nerandomilast exposure, however, a positive overall treatment impact remained. Secondary endpoints were also favourable, with mortality reductions and delayed time for supplementary oxygen requirement in the FIBRONEER-IPF study. Diarrhoea was the most common side-effect, especially when combined with nintedanib. Other adverse events noted were cough, worsening of PF, depression, anxiety, nausea, weight loss, and nasopharyngitis. Consistent results were observed in the FIBRONEER-ILD study in patients with PPF of varied subtypes of ILD.
There are a number of phase 2b RCTs currently recruiting in IPF that have shown favourable phase 2a results. These include admilparant (BMS-986278), a second-generation lysophosphatidic acid receptor 1 antagonist (LPA). LPA binding to G-protein-coupled LPA receptors drives fibroblast activation, myofibroblast differentiation, and collagen deposition. In an earlier 26-week phase 2, parallel-arm, multi-centre, double-blind, placebo-controlled RCT trial with a first-generation LPA antagonist BMS-986020 in 144 patients with IPF there was a significant slowing of FVC decline and improved quantitative lung fibrosis (QLF) scores. However, four patients developed serious adverse events, related to elevated liver function tests and gallstones, leading to study termination. The hepatotoxicity seen with BMS986020 was considered drug-specific, prompting development of a second-generation LPA1 antagonist, admilparant (BMS-986278). In a 26-week phase 2, double-blind, placebo-controlled RCT (NCT04308681) including patients with IPF (n=278 randomised, n=276 treated) and PPF (n=125 randomised, n=123 treated), admilparant reduced FVC decline compared to placebo by 1.4 per cent in IPF (95% CI: –0.1 to 3.0) and 3.2 per cent in PPF (95% CI: 0.7 to 5.7), with benefits seen irrespective of concomitant treatment. There were no concerns of hepatotoxicity and excitingly, phase 3 trials are currently recruiting.
Furthermore, evolving research helps us understand more pathways implicated in PF. The hedgehog pathway represents one such pathway activated in PF. A trial of a hedgehog inhibitor, ENV-101, found that this inhibitor increased mean FVC and decreased lung fibrosis when taken once daily for 12 weeks, compared to placebo, The ENV-IPF-101 trial (NCT04968574) was carried out across 16 sites and involved patients receiving daily treatment of 200mg ENV-101 or placebo. ENV-101 patients showed an improved FVC of 60ml after week 12, compared to a 46.8ml decrease in the placebo group. Furthermore, ENV-101 patients showed a mean improvement in per cent-predicted FVC of 1.9 per cent, compared to a 1.6 per cent decrease in placebo. No patients treated with ENV-101 experienced disease progression in this study, compared to two in the placebo group, and no serious adverse incidents were reported.
Another potential pathway implicated in fibrosis is the angiotensin-converting enzyme (ACE) pathway, and has been the focus of therapeutic studies. Buloxibutid is a selective angiotensin type 2 receptor agonist and has been shown to stabilise FVC at 24 weeks (47ml) with an improvement of 235ml at 36 weeks, in an open label study in IPF. Levels of TGF-B1, a biomarker for fibrosis, were reduced by 57 per cent by week 24, and no serious adverse events were reported. The phase 2b ASPIRE RCT is currently underway in a larger cohort of IPF patients in a placebo-controlled study.
Current oral antifibrotics are compounded by gastrointestinal adverse effects and thus research is focused on optimising the delivery and minimising adverse effects from these existing therapies. In an open label phase 1b trial of 91 IPF patients treated with inhaled pirfenidone, there was a demonstrable stabilisation of lung function on 100mg bid dose with fewer adverse effects seen in the trial compared to historical RCTs of oral pirfenidone. The limitations of the trial were its small numbers of participants and lack of placebo control. With the findings of this trial, and given that the nebulised dose of pirfenidone is equivalent to one-fifteenth of the systemic exposure of the approved oral dosage, there is hope that optimising routes of administration of drug therapies may improve treatment adherence and tolerability. The MIST study of inhaled pirfenidone versus placebo in PPF is currently recruiting (NCT06329401). Inhaled nintedanib has also shown encouraging signs in phase 1 trials, showing significantly lower systemic exposure than the traditional oral dosage.
Cough is a debilitating symptom in patients with ILD and is challenging to manage, with some disappointing negative RCTs. Two studies have shown promising early phase results. In a randomised, double-blind, placebo-controlled crossover trial of nalbuphine therapy in 41 patients with IPF there was a 75.1 per cent reduction in daytime objective cough frequency during the nalbuphine tablet treatment period as opposed to a 22.6 per cent reduction during the placebo period. Furthermore, a prospective, multicentre, randomised, double-blind, placebo-controlled, two-way crossover trial of morphine 5mg twice daily for cough in 44 patients with IPF, demonstrated that daytime cough frequency reduced from 21.6 to 12.8 coughs per hour with morphine, whereas rates did not change with placebo. Treatment adherence was 98 per cent in both groups, with a 40 per cent adverse event rate (mainly nausea and constipation) observed in the morphine group compared to 14 per cent in placebo. With misuse and physical dependence on opioids creating a barrier to their potential usage for ILD patients with chronic cough, further research on this drug class, particularly involving adverse events, is strongly welcomed.
Summary
ILDs are a diverse and complex group of disorders that require a comprehensive, multidisciplinary approach for accurate diagnosis and effective management. Advances in imaging, MDT collaboration, and emerging precision medicine tools – including biomarkers and AI – are reshaping the diagnostic landscape. While antifibrotic and immunosuppressive therapies have improved outcomes in select ILD subtypes, they remain limited in halting disease progression. Non-pharmacological interventions such as pulmonary rehabilitation and ambulatory oxygen also play a key role in symptom relief. Moving forward, personalised care strategies and broader access to MDT infrastructure will be essential to improving outcomes and reducing global disparities in ILD care.
References on request








Leave a Reply
You must be logged in to post a comment.