






Millions of people around the world suffer from the effects of a host of undiagnosed rare diseases, but a new initiative aims to decrease that number.
The US National Institutes of Health (NIH) Common Fund recently announced that the Undiagnosed Diseases Programme (UDP), a pilot programme launched in 2008, has been expanded to become the Undiagnosed Diseases Network (UDN). The Network is a federally-funded coalition of universities, clinicians, hospitals and researchers dedicated to solving the toughest medical mysteries in the US by using advances in medical technology and genetic science to help identify rare, sometimes unknown, illnesses and aims to provide answers for patients with mysterious diseases and to learn more about the disorders.
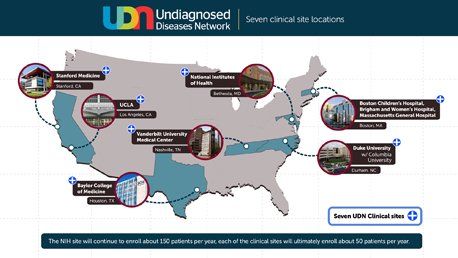
Under the initiative, seven clinical sites are now conducting scientific investigation in cases that involve patients with chronic, undiagnosed conditions. The clinical sites are located at NIH headquarters in Maryland, Harvard Teaching Hospitals, Duke University in North Carolina, Vanderbilt University Medical Centre in Tennessee, Baylor College of Medicine in Texas, the University of California, Los Angeles, and Stanford Medicine.
The UDN Co-ordinating Centre is based at the Department of Biomedical Informatics at Harvard Medical School.
The Network also consists of two sequencing core facilities, a metabolomics core, a model organisms screening centre, and a central bio-repository.
Researchers studying a patient’s gene expression and disease progression can make models, for example, using nearly-transparent zebra fish, whose genetic structure is similar to that of humans. Scientists can conduct whole-genome sequencing, which allows the medical team to read a patient’s DNA and identify changes that can reveal what may be causing a disease.
<h3 class=”subheadMIstyles”>Evolved</h3>
Ms Cecilia Esteves is the Project Manager of the UDN Co-ordinating Centre at Harvard Medical School and explained in an interview with the <strong><em>Medical Independent</em></strong> (<strong><em>MI</em></strong>) how the Network has evolved in recent years.
“The goal of the UDP was to bring individuals with mysterious medical conditions to the NIH for a week-long clinical evaluation in hopes of providing long-sought answers, while also furthering our knowledge of diseases. From May 2008 through May 2014, the UDP received nearly 10,000 inquiries, reviewed more than 3,000 applications, and accepted 750 patients into its programme.
“Building on the successes of the UDP and the lingering unmet needs of individuals and families living with undiagnosed conditions, the NIH extended the programme into a network of hospitals and laboratories across the country; the UDN.”
Patients apply to participate through an online portal called the UDN Gateway, which is maintained by the Co-ordinating Centre at Harvard Medical School. Each application is routed to a clinical site, typically the site closest to where the patient lives, Ms Esteves told <strong><em>MI</em></strong>. “The clinical site reviews the patient’s application and medical records and decides whether to offer enrollment. A network-wide review committee made up of clinicians across the UDN sites meets regularly to discuss applications and offer suggestions.
“Once accepted, patients are asked to consent to the study and travel to the UDN clinical site for an evaluation. The evaluation typically involves visits with multiple specialists, laboratory tests, specimen collection for clinical and research testing, and imaging studies. The majority of patients also undergo some level of genetic testing.”
<img src=”../attachments/dc9295f3-4def-4bfb-98f7-872323fb5c9f.JPG” alt=”” />
<strong>Ms Cecilia Esteves</strong>
A signature feature of the UDN is the compression of a patient’s clinical evaluation into the equivalent of five inpatient days, she explained, which requires rigorous coordination of activities at the clinical site. The evaluation involves detailed clinical consultations by multiple specialists, laboratory testing, diagnostic procedures and imaging studies, all tailored to the patient’s individual presentation.
<h3 class=”subheadMIstyles”>Patient stories</h3>
Ms Esteves shared with <strong><em>MI</em></strong> some of the stories of families who have sought the UDP/UDN’s help. People like Anastasia, who spent 20 years searching for a diagnosis for two of her three children but has finally learned what caused two of her children to lose their ability to speak and walk when they were just infants.
Anastasia is the mother of Amanda (21) and Daniel (15), both of whom have been affected by undiagnosed diseases. Her other son, Nick (18), is unaffected.
Amanda progressed normally for the first 10 months of her life, Anastasia told the health consumer publisher, Healthline. “She stopped reaching those milestones. She was sitting and playing with toys a little bit, babbling, drinking a bottle and finger-feeding herself. She never did crawl or walk. She could roll and pull to stand. But then all of that stopped. Over the course of a month, she lost those skills. It started with a lumpy posture and she began falling over.”
Despite visiting many different doctors over many years, including those from large academic hospitals in New York, Anastasia said, “no-one could tell us what it was. They could tell us a million things it was not. Amanda plateaued and didn’t have any motor function. She could move, but it wasn’t functional movement. It was uncontrolled. It wasn’t enough for her to complete a task. She seemed happy. I could tell that she was bright and that she could understand me as much as a baby could. I could clearly see in her eyes that she was communicating.”
Before Amanda turned three, Anastasia had a son, Nick, who does not have the condition. When she gave birth to her third child, Daniel, he progressed normally until he reached nine months old, when she started to see some regression. “We caught it early on. They admitted him to the hospital and gave him high doses of steroids and ran all kinds of tests. Technology had advanced. I was very hopeful that we would figure it out because we were catching it while it was happening. With Amanda, by the time we got to these top doctors, her regression was done. She had plateaued. Daniel was right in the middle of it when we started looking for things, but to no avail.”
It was when Anastasia’s father saw a news item featuring Dr William Gahl from the UDP that the family’s hopes were raised of finally getting a possible diagnosis.
Six weeks after sending the required information to the UDP, Anastasia received a call that Amanda and Daniel had been accepted into the programme.
The family went to the NIH in Maryland in December 2011 for one week. “We were busy from Monday through Friday from 8am to 6pm. We saw neurologists, physical therapists, speech therapists, podiatrists and genetic ophthalmologists,” Anastasia told Healthline. “They did MRIs, spinal taps, took muscle, skin, urine, and blood and x-rayed every bone in their bodies. They also took blood and urine from me, their father and my other son,” she said.
Before the family went home, Dr Gahl told them he was confident that someday they would be able to offer a diagnosis. That day came the following September, when the family was called back to the NIH to discuss the findings. It was then they found out their children had Aicardi-Goutieres syndrome (AGS) 2B.
“We were told there are several mutations of this gene and ‘your children have 2B’. My husband and I are carriers. With this syndrome, if you have one good copy of this gene, you’ll function fine. But if you have two bad copies, you have Aicardi-Goutieres 2B. My husband and I each had a good copy and a bad copy, and when Amanda and Daniel were conceived, we passed our bad copies to them.”
This neurological disease occurs in all populations worldwide. It is believed to be under-diagnosed but there are at least 450 cases of AGS globally.
“I had never met anybody with two kids with something as severe as my kids. When they told me what they had, and there were other families, some like mine, some with three kids affected, and worldwide, I was overwhelmed with emotion,” Anastasia said.
“I just started to cry. I wasn’t happy or sad, it was just relief. I was on cloud nine — ‘I have a diagnosis’. I don’t think we ever would have found out if we didn’t have someone looking at their genetic make-up. I’m very grateful for the programme. For almost two decades, we didn’t know.”
Today, Daniel is in high school and Amanda has just finished school. She works at a day care nursery school, programming her computer to speak out instructions for kids to do arts and crafts. She is also a recreation co-ordinator, creating flyers detailing daily activities for retirees.
Then there is the ongoing case of Andrew (28). His nightmare began at the age of 19 when he started trembling and couldn’t speak. Doctors suspected he was suffering from anxiety and prescribed medication to control it. But that didn’t help. He suffers seizures that leave him temporarily paralysed and he can no longer feed himself or walk on his own.
The past nine years have been a blur of doctors’ appointments, hospital visits and medical tests that have failed to produce answers. The family visited several hospitals. Doctors discovered that the receptors in his brain were malfunctioning and that he lacked sufficient dopamine. As a result, Andrew has some symptoms similar to those of Parkinson’s disease, yet his symptoms did not add up to any known disease.
His family decided to seek the help of the UDN at its site at the University of California in Los Angeles (UCLA).
One of UCLA’s principal investigators and the lead doctor on Andrew’s case, Prof Stanley Nelson, explained the UDN can examine all known genes, not just the ones believed to have mutations that cause diseases, and doing that can lead to the discovery of new illnesses.
“We have powerful techniques to look at every gene that is being expressed, as well as every gene that is inherited. This is an example of true precision medicine,” Prof Nelson told <em>Kaiser Health News</em>. “Part of what we have to do is keep building that library, that encyclopedia of what gene and what gene mutations cause what symptoms. It’s just incomplete at this moment.”
Andrew underwent several diagnostic and other tests, including a muscle biopsy, an EEG, MRI and a lumbar puncture. He also met with UCLA specialists in brain degeneration and muscle and nerve disorders.
Prof Nelson then sat down with the family to explain what he had found. He had reviewed Andrew’s genome and compared it with that of both parents. Andrew had one copy of a defective gene that leads to Parkinson’s but the genome sequencing did not show a second copy, without which it could not be Parkinson’s.
He explained that Andrew’s illness was clearly progressive and that his brain was shrinking, making it harder for him to process language and information. He said he still did not have a diagnosis but believed it was a new disease.
Prof Nelson is continuing to pore over the test results, conducting additional exams and communicating with others in the UDN. So far, no complete answers have been found but Andrew’s family is hopeful that the Network will make further progress over time in unravelling the mystery of his illness.
<h3 class=”subheadMIstyles”>Huge potential</h3>
Among those diagnosed at the UDP from 2008 to 2015, the length of time to arrive at a diagnosis after the on-site evaluation ranged from one week to four years.
The UDN began accepting applications in September 2015, and “since then, we have received more than 1,800 applications and accepted nearly 800 patients into our programme. Collectively, the seven UDN clinical sites have evaluated more than 500 patients and made some 140 diagnoses. These diagnoses include several new conditions and new manifestations of known conditions. Several UDN case reports have been published in the scientific literature,” Ms Esteves told <strong><em>MI</em></strong>.
She believes the UDN also holds huge potential for new medical discoveries. The large number of patients with rare and newly-recognised conditions, the breadth of expertise in the network and the collaborative nature of the UDN, she said, represent an opportunity to accelerate discoveries about health and disease, while developing best practices for the diagnosis of challenging cases.
It is a view that is shared by the <em>American Journal of Human Genetics</em> (<em>AJHG</em>), which has hailed the work of the UDN and its contribution to medicine. “Building the UDN has already brought insights to human and medical geneticists. The initial focus has been on establishing common protocols for institutional review boards and data sharing, creating protocols for referring and evaluating patients, and providing DNA sequencing, metabolomic analysis, and functional studies in model organisms.”
The journal suggests the UDN model could be extended around the US to help improve difficult diagnoses. “By extending this precision diagnostic model nationally, we strive to meld clinical and research objectives, improve patient outcomes and contribute to medical science. Long diagnostic odysseys are expensive and they involve repeated and duplicative diagnostic efforts with risks of invasive procedures and false diagnoses.
“The UDN was designed to forge a paradigm for diagnosing rare and previously unrecognised diseases, improving the management of these conditions and accelerating research on rare diseases. The UDN exists to increase the national capacity to assess those with mysterious medical conditions, to create a network with broad collaborative expertise and to determine ways to extend this model to other medical centres.”
UDN researchers and doctors say the lessons that the network holds for medicine extend well beyond the individuals evaluated. The overwhelming majority of UDN patients come to the network after misdiagnoses or no unifying diagnosis at all. In the general population, many common diseases, such as cardiovascular conditions, cancer, and infections — not just rare diseases — are often misdiagnosed. Most Americans experience a diagnostic error — a delayed diagnosis, missed diagnosis or misdiagnosis — which can cause harm because of delayed or inappropriate tests and treatments.
The work of the UDN can make a major contribution to medicine through its focus on rare or unknown diseases by bonding scientists and clinicians to study diseases by exploring their causes at the clinical, genetic and cellular levels, according to three leading US clinicians, Drs William A Gahl, Cornelius F Boerkoel and Manfred Boehm, all of whom are familiar with the work of the UDN.
Writing in the online biomedical research journal <em>Disease Models & Mechanisms</em>, they said some of the most remarkable discoveries in physiology and medicine are uncovered by studying rare conditions because the importance of certain molecular mechanisms is revealed only when their dysfunction results in disease. “The UDP has enabled the discovery of several new diseases and disease mechanisms through collaborations between clinical and basic science teams, using the power of both clinical medicine and biological models.”
They suggest this approach could work in establishing programmes with similar infrastructure at other centres around the world that could benefit patients, their families and the entire medical community by enhancing research for rare and novel diseases.
Indeed, a number of international sites have expressed interest in learning from the UDN experience. As a result of two international conferences, one in Rome in 2014 and another in Budapest in 2015, the Undiagnosed Diseases Network International (UDNI) was established. Its board of directors reflects its international character — it includes experts from Australia, Canada, Hungary, Italy, Japan and the US.
<h3 class=”subheadMIstyles”>Challenges</h3>
But even as the UDN extends its mission globally, it still faces a number of challenges, Ms Esteves told <strong><em>MI</em></strong>. “Aside from the obvious diagnostic challenges, one of the most significant challenges of our work is finding additional patients to confirm suspected diagnoses.
“This challenge is not unique to the UDN. It is part of the daily life of anyone working in rare disease research. To address this challenge, we aim to collect and share our data as widely and responsibly as possible. But that alone isn’t enough. We also need to rely on others to share their data. Fortunately, a lot of progress has been made in this area over the past few years through data-sharing initiatives like Matchmaker Exchange (MME).”
The MME project was launched in October 2013 to focus on the simple matching of unsolved rare disease cases that share a common phenotype and candidate gene. The MME aims to expand the scope of analysis to genes and genomic variation already implicated in genetic disorders. Another goal is to more effectively support the role of patient-initiated matchmaking in the MME. It also enables investigators with unsolved rare disease cases to submit their patient data and thereby find each other and undertake selective data-sharing.
The UDN also faces concerns about funding cuts that could imperil its work. The UDP was initially funded by the NIH Office of Rare Diseases Research. It secured a five-year budget of $120 million from the NIH Common Fund, with the National Human Genome Research Institute (NHGRI) serving as the lead administrator. In 2017, the NIH Common Fund approved funding for the second phase of the UDN, Ms Esteves told <strong><em>MI</em></strong>.
Despite securing such funding, however, there is concern among some that a proposal by President Donald Trump to cut the NIH budget by $5.8 billion could put some of the work of this ground-breaking network in jeopardy at a time when it is producing tangible results and bringing hope to vulnerable patients and their families.
<div style=”background: #e8edf0; padding: 10px 15px; margin-bottom: 15px;”> <h3 class=”subheadMIstyles”>Patients seeking a diagnosis</h3>
These are profiles of some of the patients seeking help from the UDN.
<p class=”subheadMIstyles”><strong>Participant 039</strong>
Male, aged 12 years, with overgrowth and developmental delay caused by a change in the GNAS gene.
After birth, the patient was found to be in respiratory distress. He had low levels of red blood cells (anaemia), yellowing of the skin (jaundice caused by hyperbilirubinaemia), and heartburn (gastro-oesophageal reflux). This required hospitalisation in the NICU for 72 days.
Over time, the patient was noticed to have delayed milestones (global developmental delay), walking and talking at approximately two years of age. He progressed quickly afterwards. However, he still has difficulty walking and currently uses a walker and sometimes a wheelchair. He experiences painful bone cracking and hand, foot, and ankle discomfort when walking.
At the age of nine, he was seen by endocrinology due to increased height and weight, despite diet and exercise. Currently, he continues to grow at a fast pace, with his clothing and shoe size going up over the span of just a few months.
<p class=”subheadMIstyles”><strong>Participant 038</strong>
Female, aged three years, with pontocerebellar hypoplasia, type 2D.
At birth, the patient had low muscle tone (hypotonia) and poor head control. She was initially able to breast-feed, but stopped at the age of seven months. At eight months, an MRI showed abnormal brain findings (cerebellar atrophy). She was admitted to the hospital at the age of 10 months for sleep problems and abnormal breathing (stridor).
Currently, the patient has developmental delay and still struggles with head control. She does recognise her mother and has a social smile, but appears to have vision impairment (cortical visual impairment). She also sweats a lot and needs a G tube for feeding. At times, she has low blood glucose levels (hypoglycaemia). She has had seizures that are being controlled by medication.
<p class=”subheadMIstyles”><strong>Participant 034</strong>
Female, aged 26 years, with muscle weakness and pain (myalgia), chronic fatigue, exercise intolerance and severe episodes of weakness, dizziness and paralysis.
When the participant was nine years old, she started having episodes of muscle weakness, myalgia, nausea and dizziness. Other symptoms during severe episodes include paralysis, feeling cold, slow heart rate and low blood oxygen levels. These episodes would range from several minutes to several days. The episodes are typically triggered by physical activity, fasting, fatigue, stress and extreme temperatures.
At the age of 19, the participant’s symptoms got much worse. She began to experience severe fatigue and weakness, which affected her ability to move and exercise. She was also found to have difficulty walking (scissor gait, gait disturbance, gait ataxia), GI issues (gastrointestinal dysmotility), cold and heat intolerance, excessive sweating (hyperhidrosis) and exercise intolerance. At 24, she was diagnosed with postural orthostatic tachycardia syndrome (POTS).
<p class=”subheadMIstyles”><strong>Participant 030</strong>
Male, aged 70 years, with Schnitzler syndrome.
Around age 60, the patient started having recurrent episodic fevers, with associated muscle/joint pain and a hive-like rash across his upper chest and abdomen that would come and go with the fevers. He had extensive evaluation for occult infection or malignancy that was unrevealing. He was trialled on numerous immunosuppressive regimens. At present, with his diagnosis of Schnitzler syndrome, he is on tocilizumab and prednisone.
<p class=”subheadMIstyles”><strong>Participant 020</strong>
Female, aged 44 with history of migraine headaches, sleep apnoea, anxiety, myoclonic jerks, temperature intolerance, arthritis of the hands and osteoarthritis of the spine.
This patient has had constant headaches for over 20 years. At age 34, she gave birth to her daughter and experienced intense problems with temperature regulation, feeling intense burning on her skin and within her whole body. She had a fall at age 22 and began to experience spine troubles, but the symptoms worsened quickly around age 38 and she had hip surgery at age 40. Around age 42, she began experiencing jerking of her arms and legs, especially when falling asleep, waking up, or relaxing. Around this same time she also began experiencing sleep apnoea, which gets better with the use of a CPAP (continuous positive airway pressure) machine. She notes occasional trouble retaining short-term memory. Around age 40, she developed arthritis in her fingers.
<em>Participant profiles used with permission from the US Undiagnosed Diseases Network.</em>
</div>












Leave a Reply
You must be logged in to post a comment.