






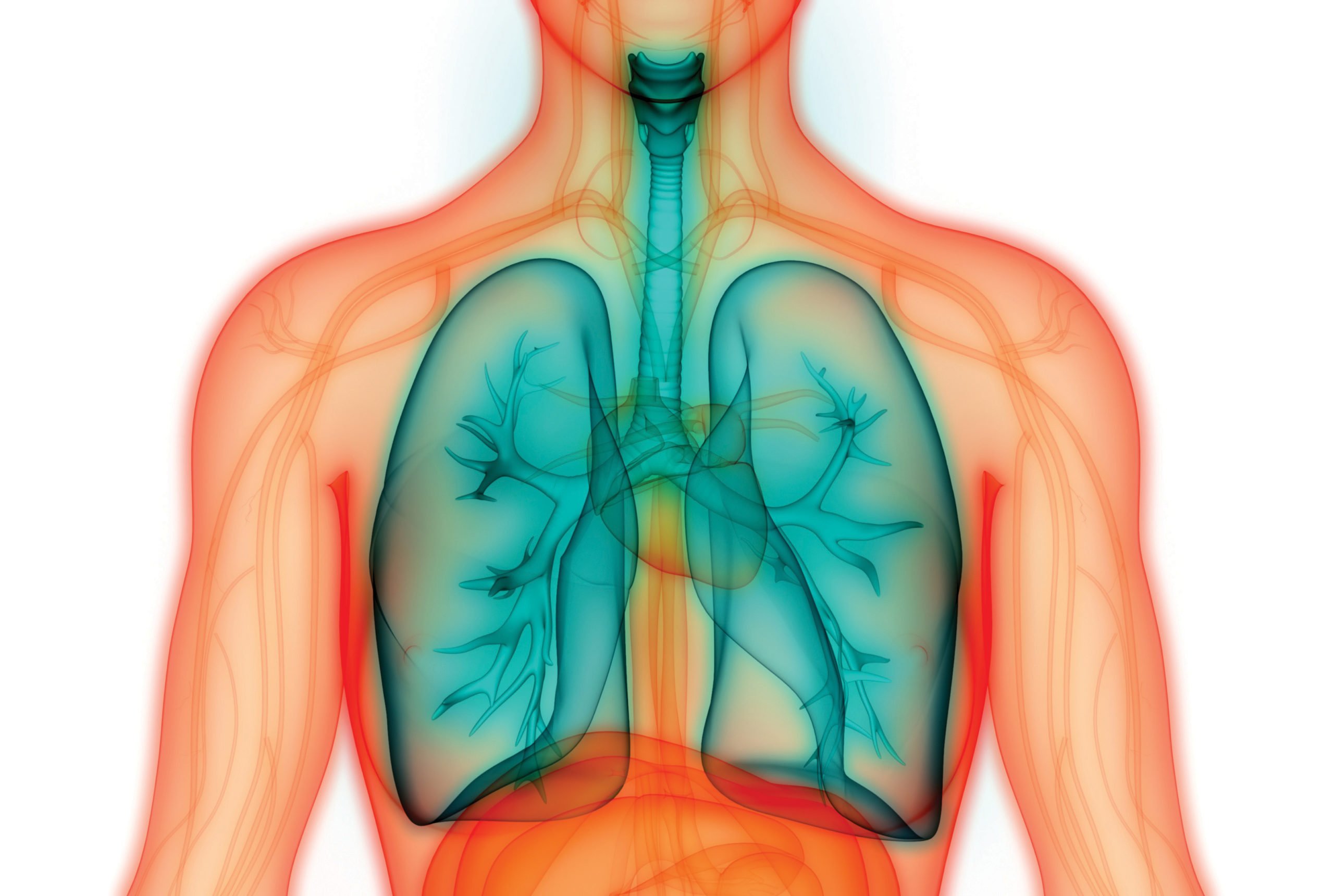
Cystic fibrosis (CF) is an inherited chronic disease, primarily affecting the lungs and digestive system. This lifelong condition usually becomes more severe with age and affects both males and females in equal proportions. The symptoms and severity of CF vary from person to person, but the majority of people with the disease have both respiratory and digestive problems. There is no cure for CF, however, life expectancy has increased steadily over the past 20 years due to better disease understanding, treatment advances, and earlier diagnosis.
Pathophysiology
CF is caused by a gene mutation leading to dysfunction of the cystic fibrosis transmembrane conductance regulator (CFTR) protein, a chloride channel of exocrine glands. The defect leads to diminished chloride secretion, which in turn leads to increased sodium absorption through epithelial sodium channels and removal of water from secretions, which become abnormally viscous.
Essentially this causes the body to produce unusually thick sticky mucus that clogs the lungs and obstructs the pancreas, stopping natural enzymes from enabling the body to break down and absorb food. The effects cause obstruction, inflammation, infection in the lungs and upper airways, tissue reorganisation, and loss of function.
CF affects multiple organ systems including the lungs, pancreas, upper airways, liver, intestine, and reproductive organs to varying degrees. The severity of the disease in the individual depends on variable organ sensitivity and on the genetically determined residual function of the CFTR protein. Almost all (97-98 per cent) affected male patients are infertile because of obstructive azoospermia and 87 per cent of patients have exocrine pancreatic insufficiency. Disease severity, particularly the degree of pulmonary involvement, also depends on other disease-modifying genes and a number of factors including socioeconomic background.
Symptoms
People with CF present with a variety of symptoms, including very salty-tasting skin, persistent coughing with phlegm, frequent lung infections, wheezing or shortness of breath, poor growth/weight gain despite a good appetite and frequent greasy, bulky stools or difficulty in bowel movements.
Symptoms usually appear in the first year of life, although occasionally they can develop later. The thick, viscous mucus in the body affects a number of organs, particularly the lungs and digestive system. Typical respiratory symptoms include cough and wheeze. Recurring chest and lung infections are caused by the continual build-up of mucus in the lungs, which provides an ideal breeding ground for bacteria.
Diagnosis
Early diagnosis provides opportunities for earlier medical intervention and improves the quality- and length-of-life of CF patients. Providing infants with the best possible care results in better nutritional and lung function outcomes later in life.
Chronic disease of the lungs and paranasal sinuses varies among patients with CF and can be difficult to distinguish from frequent recurrent bouts of bronchitis and/or pneumonia, especially in young children. Children with cough, sputum production, or wheezing of more than three months’ duration, persistently abnormal radiological findings, persistently positive bacterial cultures of respiratory secretions, or clubbing of the fingers should undergo diagnostic testing for CF even if their neonatal screening test was negative. The same applies for children with bilateral chronic rhinosinusitis with frequent exacerbations, with or without nasal polyps.
Most cases of CF are diagnosed through screening tests, which are carried out early in life. However, some babies, children, and even young adults are diagnosed later following unexplained illness. The main ways of diagnosing CF are newborn screening (heel prick test), antenatal testing (chorionic villus sampling (CVS)), carrier testing (genetic screening), and sweat testing. If both parents are carriers of the faulty gene, but don’t have CF, a child has a one-in-four chance of being born with CF; a two-in-four chance of being a carrier, but not having the condition; and a one-in-four chance of being completely free of the condition.
About 2,000 CFTR gene mutations have been identified to-date and genetic testing provides important information to assist the diagnosis and treatment of CF. F508del is the most commonly detected CFTR mutation in Irish people living with CF. In 2016, 91.6 per cent of the CF population had at least one copy of F508del, 15.2 per cent had at least one copy of G551D, and 5.8 per cent had at least one copy of R117H. All other CFTR mutations affect less than 5 per cent of the CF population. Just over 55 per cent of patients living with CF in Ireland in 2016 had two copies of the F508del mutation (F508del homozygous). Approximately one-in-19 of the indigenous population are thought to carry one copy of the altered gene that causes CF. Testing is available at the National Centre for Medical Genetics in Crumlin, Dublin.
Associated health issues
Organisms detected in the airways of individuals with CF in Ireland in 2016 included Staphylococcus aureus (54.7 per cent) and Haemophilus influenza (29.0 per cent), found most frequently in children. Pseudomonas aeruginosa was detected in at least one respiratory sample of 60.3 per cent of adults.
Two strains of bacteria that commonly infect people with CF are Pseudomonas aeruginosa and Burkholderia cepacia complex. They multiply in the thick mucus inside the lungs and may cause serious health problems and repeated chest infections.
As more and more people with CF become infected with these bacteria, antibiotic resistance is a growing issue, which is why cross-infection is such a problem. Lung infections caused by Pseudomonas aeruginosa and Staphylococcus aureus can become chronic and cause an irreversible reduction in lung function.
CF causes mucus to block the ducts in the pancreas leading to malnutrition, steatorrhoea, and diabetes. People with CF who develop diabetes may find it difficult to gain weight or may lose weight and see a decline in their lung function. CF-related diabetes is usually controlled by regular injections of insulin. Diabetes rarely occurs in children with CF.
Pancreatic insufficiency (PI) is manifested by voluminous, fatty, shiny, malodorous, pulp-like stools, abdominal symptoms, dystrophy, and deficiencies of fat-soluble vitamins (eg, haemolytic anaemia due to vitamin E deficiency) and trace elements (eg, zinc dermatosis). The diagnosis can be established by a low faecal elastase measurement. Patients with primary pancreatic insufficiency are at increased risk of chronic and/or recurrent pancreatitis.
People with CF are also prone to sinusitis and hay fever. Older children with CF can develop a form of arthritis in joints such as the knee, which improve with time and treatment. Older children and adults with CF are also prone to osteoporosis due to repeated infection, poor growth or weight, lack of physical activity and lack of vitamins and minerals due to digestive problems, with steroid use also a risk-factor.
One-in-ten babies with CF is born with meconium ileus and an operation to remove the blockage will probably be required.
In nearly all men with CF, the spermatic tubes do not develop correctly, making them infertile.
Women with CF may find their menstrual cycle becomes absent or irregular if they are underweight. There is also an increased thickness of cervical mucus, which may reduce fertility. However, most women with CF can conceive without any difficulty. If the bile ducts in the liver become blocked by mucus as the disease progresses, this can be serious and in some cases a liver transplant may be necessary.
Treatment
There remains no cure for CF. Improved diagnosis and symptomatic treatments have increased the survival rates of people with the condition, however, from a life span of only a few months in the 1950s to a global median age of 40 years today. The predicted median age of survival for a person in Ireland with CF is early to mid-30s. Aside from symptomatic treatment, the first mutation-specific treatments have recently become available.
The aim of symptomatic treatment is to clear and control infections in the lungs and digestive system and reduce long-term damage caused by infection and organ complications.
Inhaled bronchodilator drugs are used to relax the muscles surrounding the airways and help the person breathe more easily. Antibiotics are prescribed to treat infections in the lungs. All young children diagnosed with CF are started on a course of oral antibiotics to protect them from certain bacteria, which will be continued until they are three years of age. Steroids are used to help reduce the swelling of the airways, which can help with breathing and DNase, an enzyme, which is usually inhaled, is used to help thin and break down the viscous mucus in the lungs, so it is easier to expectorate.
Pancreatic enzyme replacement therapy (PERT) is required by almost all individuals with CF, due to pancreatic insufficiency. When the exocrine pancreas no longer functions adequately, the production of pancreatic enzymes is reduced, leading to fat malabsorption. The consequences of this include steatorrhoea, malnutrition, and fat-soluble vitamin deficiencies. The vast majority (91 per cent) of patients with CF in Ireland received PERT in 2016. Supplementary feeding can be required due to continued weight loss. Nearly 38 per cent of individuals with CF in Ireland required supplemental feeding in 2016. Approaches to supplemental feedings included oral supplementation, gastrostomy tube/button feeding, nasogastric tube feeding, and total parenteral nutrition.
Pancreatic enzymes are taken before and during every meal and fat-containing snack or drink, helping the digestive system break down food so that it can be digested and absorbed. The number of enzyme capsules taken needs to be adjusted depending on the amount of fat in the meal, snack, or drink. The enzymes should be taken with the meal and the timing may vary depending on the age of the person with CF. It is essential that people with CF receive advice about enzymes from a specialist CF dietitian. Fat-soluble vitamin supplements (A,D,E, and K) are taken to help replace lost vitamins and to prevent deficiencies. Because people with CF lose fat in their stools, they also lose the fat-soluble vitamins. Nutritional supplements can help compensate for poor digestion and provide additional energy and nutrients.
Bisphosphonates are taken to treat osteoporosis, which can occur as a result of CF, to help maintain bone density and reduce the risk of fractures.
It is important that people with CF are up-to-date with all the required vaccinations and should have the flu vaccine annually, as they are more susceptible to complications as a result of infection.
Daily physiotherapy is usually required and if the person with CF has a chest infection the amount of airway clearance will need to be increased. Physiotherapy is tailored to the individual’s needs. While physiotherapy for CF mainly focuses on airway clearance, its role has expanded to include daily exercise, inhalation therapy, posture awareness, and in some cases for the management of incontinence.
In severe cases of CF, when the lungs fail, a double lung transplant may be required. Lung transplantation for CF has a good success rate: 70 per cent of patients survive one or two years after transplantation and the longest surviving patients had their transplant operation over 15 years ago.
CFTR potentiators and correctors used in the treatment of CF: CFTR potentiators are the first available treatments that target the defective CFTR protein, the underlying cause of cystic fibrosis. CFTR correctors (elexacaftor, lumacaftor, tezacaftor) correct the processing and trafficking defect of the F508del-CFTR protein to enable it to reach the cell surface where the CFTR potentiator, ivacaftor, can further enhance the ion channel function of the CFTR protein (See Table 1).
Trikafta: New treatment for CF to become available in
Ireland in 2020: Trikafta, the first drug to treat the underlying cause of CF, was recently approved for cover by the HSE – before it received European Medicines Agency (EMA) approval. The HSE announced in December 2019 that it had reached agreement with the manufacturer, Vertex Pharmaceuticals, to expand its existing reimbursement agreement to include Vertex’s first triple combination therapy Elexacaftor (VX-455), Tezacaftor, and Ivacaftor (Kalydeco) – known as Trikafta for Irish CF patients. The agreement with Vertex arises from the 2017 pipeline agreement with the company, which provided funding for existing products as well as new ones then in development. Trikafta is the first approved treatment that is effective for CF patients aged 12 years and over with at least one F508del mutation, which affects 90 per cent of the population with CF worldwide and 80 per cent of the CF population in Ireland. All CF patients in Ireland who are aged 12 years and over and who have at least one copy of the F508del gene mutation will get access to Trikafta as soon as it is approved by the EMA. While a decision by the EMA was originally expected to be made in the first quarter of 2020, with the drug becoming available by June, Cystic Fibrosis Ireland said it predicts that the decision is now more likely in the autumn/winter of 2020, with the likely reason for non-fast tracking of Trikafta because it would be important that the EMA license for Trikafta is as broad as possible, ie, covering as many altered gene combinations as possible.
Once approved by the EMA, it is anticipated that most patients on existing CF drugs such as Orkambi will transfer over to Trikafta depending on the advice of their clinician. One clinical trial has shown Trikafta to increase lung function by a mean of 14 per cent, while other research has shown its use to cause a decrease in exacerbations by up to 60 per cent. Commenting on the development, Mr Philip Watt, CEO of Cystic Fibrosis Ireland, said: “The decision effectively means Ireland will be the first country in Europe to receive this drug for its patients. We have great hope now that the life expectancy of people with cystic fibrosis in Ireland will increase steadily over the next few years and, indeed, Ireland may overtake other countries.”
While news of this new drug is very welcome, about 10 per cent of CF patients remain without an available corrective therapy, since they do not make any CFTR protein.
Cystic Fibrosis Ireland Annual Conference
Dr Preston Campbell, former president and chief executive officer of the Cystic Fibrosis Foundation in the US, and a leading medical expert in CF, has been announced as the keynote speaker at the forthcoming Cystic Fibrosis Ireland annual conference taking place in Galway from Friday 3 April – Sunday 5 April. Full information at www.cfireland.ie. The conference comes ahead of Cystic Fibrosis Ireland’s annual flagship fundraising appeal, 65 Roses Day, on Friday, 10 April, where people can lend their support by buying a purple rose for €2 or by donating online at www.65RosesDay.ie.
References on request
Ivacaftor (Kalydeco)
Ivacaftor (Kalydeco) potentiates the CFTR protein, a chloride channel present at the surface of epithelial cells in multiple organs. This facilitates increased chloride transport by potentiating the channel-open probability of certain CFTR gene mutations. It is indicated for CF in adults and children aged six months or older who have one mutation in the CFTR gene. It is not effective when used without a CFTR corrector (eg, lumacaftor) if the patient is homozygous for the F508del mutation in the CFTR gene.
Lumacaftor/ivacaftor (Orkambi)
This combination product contains lumacaftor, a CFTR corrector. Lumacaftor corrects the processing and trafficking defect of the F508del-CFTR protein to enable it to reach the cell surface where the CFTR potentiator, ivacaftor, can further enhance the ion channel function of the CFTR protein. Ivacaftor facilitates increased chloride transport by potentiating the channel-open probability of the CFTR proteins. The combination is indicated for CF in patients aged six years or older who are homozygous for the F508del mutation in the CFTR gene. This combination is well-tolerated in young children.
Tezacaftor/ivacaftor (Symdeko/Symkevi)
CFTR corrector and potentiator combination regimen. It is indicated for CF in patients aged ≥6 years who are homozygous for the F508del mutation or who have at least one mutation in the CFTR gene that is responsive to tezacaftor/ivacaftor based on in vitro data and/or clinical evidence.
Table 1: CFTR potentiators and correctors
CF in Ireland
Ireland has the highest incidence per head of population of CF in the world, with three times the rate of the US and the rest of the European Union. Between 2008 and 2016 an average of 38 individuals in Ireland were diagnosed with CF each year, with 22 diagnosed in 2016.
At the beginning of each year, the CF Registry of Ireland conducts a survey of hospitals known to provide care to people with CF in the Republic of Ireland. The registry has gathered information on people with CF since 2002. The annual hospital census carried out by the registry in 2016 showed that 1,339 people were living with CF in Ireland. Most patients attended one hospital for CF care (n=1,107, 82.7 per cent), whereas 17.3 per cent (232) shared care at two or more hospitals. The year 2016 was also the first time that the median age for people with CF reached 20 years (half of the population were aged over 20 years and half under) in Ireland, while 8.9 per cent of the CF population in Ireland was recorded as over 40 years old. There were 1,266 individuals (565 children and 701 adults) with CF on the registry on the last day of 2016, representing 94.5 per cent of people living with CF in Ireland in that year.
Over half of individuals with CF in 2016 were located in Leinster, approximately a third in Munster, and the remainder (14.6 per cent) in Ulster and Connaught. The counties with the largest numbers of individuals included Dublin (n=354), Cork (n=165), Limerick (n=76), Galway (n=59) and Tipperary (n=59), and more males (58.0 per cent) than females (42.0 per cent) were recorded living with CF.
The number of people (children and adults) diagnosed in 2016 (n=22) and 2015 (n=28) is likely to be an underestimate, resulting from a delay in joining the registry. Since the introduction of the National Newborn Bloodspot Screening Programme (NNBSP) in July 2011, 156 individuals have been diagnosed as a result of the screening programme. Sixteen of the 22 new CF diagnoses made in 2016 (72.7 per cent) were made following newborn screening.
Hospitalisations recorded by the registry reflect admission for a variety of reasons, both CF and non-CF related. In 2016, 954 hospitalisations were recorded in 497 individuals. Over 70 per cent of hospitalisations (71.3 per cent) were for the purpose of treating a pulmonary exacerbation. A total 363 individuals, 32.1 per cent of individuals attending CF centres/clinics in that year, were hospitalised for treatment of a pulmonary exacerbation; 20.8 per cent of children (n=106) and 41.1 per cent of adults (n=257). Of those hospitalised for treatment of a pulmonary exacerbation in 2016, 44.4 per cent (n=161) were admitted to hospital two or more times in the year. The cumulative total number of days spent in hospital by individuals with CF for treatment of a pulmonary exacerbation in 2016 was 9,278 days; 1,689 days for paediatric patients, 7,589 for adult patients.
The number of days spent on IV antibiotics for pulmonary exacerbation for all people with CF in Ireland in 2016 came to a total of 14,084. The mean duration of pulmonary exacerbation treatment was 16.4 days and median duration was 14 days.
The registry recorded 6,792 outpatient visits to CF specialist centres and CF clinics across the country by 1,133 individuals with CF in 2016, however, CF day-unit and drop-in visit statistics are thought to be underestimated.
The number of deaths from CF in Ireland varies from year to year. Since 2000, an average of 18 individuals died annually (range: Seven to 31). The deaths of 13 people with CF occurred in 2016 (nine female, four male). These individuals died between the ages of 11 and 59 years, with the median age of death 32.5 years. Deaths were due to respiratory/cardio-pulmonary causes (n=9), transplant-related issues (n=2) and the cause of death in two individuals was not known to the registry at the time of reporting.






Leave a Reply
You must be logged in to post a comment.