NOTE: By submitting this form and registering with us, you are providing us with permission to store your personal data and the record of your registration. In addition, registration with the Medical Independent includes granting consent for the delivery of that additional professional content and targeted ads, and the cookies required to deliver same. View our Privacy Policy and Cookie Notice for further details.
Chronic lymphocytic leukaemia: Latest approaches to diagnosis and treatment
By
Fidelma Hackett
- 27th May 2024
This CPD module gives a detailed overview of the presentation, diagnosis, and latest treatment options for CLL in Ireland
To complete this module and earn free CPD points, go to www.doctorCPD.ie and answer the 10 true or false questions and complete the five MCQs based on this article.
Chronic lymphocytic leukaemia (CLL) is a chronic lymphoproliferative disorder characterised by progressive accumulation of monoclonal B-lymphocytes within the bone marrow, blood, and lymph nodes. The mature B-cell neoplasm primarily affects essential sites such as peripheral blood, bone marrow, spleen, and lymph nodes.
CLL is considered identical to small lymphocytic leukaemia (SLL), classified as an indolent non-Hodgkin lymphoma. The term CLL is used when the disease manifests primarily in the peripheral blood whereas the term SLL is used when nodal involvement is predominant.
Epidemiology
CLL is the commonest type of leukaemia in adults in the Western world, exhibiting a variable national incidence dependant on the ethnic and age distribution within the population. According to the National Cancer Registry of Ireland (NCRI), the incidence of CLL in the Republic of Ireland (ROI) is reported at six1 cases per 100,000 population.1 A recent study, focused on consecutive newly-diagnosed CLL cases documented by a centralised flow cytometry service, has indicated a higher CLL incidence in the Republic.2 This finding suggests a potential exclusion of individuals within the ‘watch and wait’ group, the ageing population of the Republic, and those aged >65 years (mainly Caucasian). CLL predominantly affects older adults with an average age at diagnosis of 70 years. The incidence rises steadily with increasing age.
Clinical presentation
The diagnosis of CLL is often arrived at incidentally during routine full blood count (FBC) examinations conducted for unrelated medical purposes. The second most common presentation involves lymphadenopathy, usually a painless swelling of lymph nodes, often in the cervical area. Less frequently, 5-to-10 per cent of patients present with B symptoms including fevers, drenching night sweats, and unintentional weight loss. Occasionally, auto-immune complications such as haemolytic anaemia, thrombocytopaenia or red cell aplasia may manifest as presenting features.
CLL has a highly variable clinical course, with up to one-third of patients not requiring treatment. The survival spectrum ranges from two-to-20 years, with a median overall survival of 10 years. This heterogeneity underscores the diverse nature of CLL presentations and the importance of comprehensive clinical assessment for tailored patient management.
Pathogenesis and risk factors
CLL arises from one or more acquired mutations to the genetic material of a single bone marrow cell, which under normal circumstances would undergo development into a healthy lymphocyte. The precise aetiology of CLL is unknown, but specific factors have been recognised as contributors to an increased risk of CLL development.
1. Increasing age: The risk of developing CLL exhibits a positive correlation with age, reaching its peak incidence between 60 and 80 years. Only 10 per cent of patients with CLL are <50 years, and occurrence below the age of 40 years is rare. CLL is notably uncommon in children.3
2. Gender: CLL affects males more frequently than females, with men being approximately twice as likely as women to develop the condition.
3. Ethnicity: The incidence of CLL demonstrates geographic and racial variation. It is most commonly observed in white Caucasian adults within Western populations, and there is a higher prevalence amongst Jews of Eastern European descent. While CLL incidence in Europe is similar to that in the US, considerable lower rates are observed in Asian countries such as Japan and China, as well as in the African-American population.
4. Family history: CLL has one of the strongest familial inherited predispositions among haematological malignancies. Approximately 10 per cent of CLL patients have a family history of the disease.4
5. Monoclonal B-cell lymphocytosis (MBL): MBL, characterised by an increased number of monoclonal B-cells, has been observed to progress to CLL, requiring treatment at a rate of 1-to-2 per cent per year.
Diagnosis and evaluation
The diagnosis of CLL is established through a combination of lymphocyte morphology, the presence of ≥5 × 109/L B lymphocytes in the peripheral blood persisting for three months, and a characteristic immunophenotype.5
1. Peripheral blood assessment:
The diagnosis of CLL is suspected when an absolute lymphocytosis is evident in the peripheral blood. FBC, including a differential white blood cell count, is essential to confirm lymphocytosis. A confirmed diagnosis of CLL requires the sustained presence of ≥5 × 109/L B lymphocytes in the peripheral blood, for at least three months.
2. Morphological evaluation:
A peripheral blood film is required to evaluate malignant cell morphology. Smear or smudge cells, characteristic of CLL, are typically observed. CLL cells manifest as small, round lymphocytes with a mature appearance, featuring a narrow cytoplasmic border, a dense nucleus devoid of discernible nucleoli, and condensed ‘cracked mud’ clumped chromatin.
3. Immunophenotype analysis:
Confirmation of CLL involves immunophenotype analysis of peripheral blood lymphocytes through peripheral blood flow cytometry. CLL cells exhibit co-expression of the surface antigen CD5 along with the B-cell antigens CD19, CD20, and CD23. The levels of surface immunoglobulin, CD20, and CD79b are characteristically low compared with those found on normal B-cells.
4. Bone marrow assessment:
Bone marrow aspirate and biopsy is not required for the diagnosis of CLL, but may be indicated clinically in specific cases such as clinical trial enrolment, cytopaenia or disease transformation.5 These procedures are typically reserved for situations where additional insights into disease progression or complications are required.
Prognostic and predictive markers are measurable in CLL, which help predict the disease prognosis and likelihood of treatment response. Several analyses are available to discern crucial molecular and genetic characteristics.
1. Fluorescence in situ hybridisation (FISH)
FISH is a highly sensitive test that detects chromosomal abnormalities in patients with CLL and can be performed with peripheral blood lymphocytes. It is required to detect deletion of chromosome 17 (del17p), and gene sequencing to identify mutations of TP53. Identification of del17p or the presence of TP53 mutation is key in guiding treatment decisions and helps inform prognosis. Common deletions include those in the long arm of chromosome 13 (del13q), trisomy of chromosome 12, and deletions in the long arm of chromosomes 11 (del11q). Given the potential evolution of genetic changes throughout the disease process, FISH analysis should be carried out as close as possible to initiation of therapy, preferably within a timeframe of less than six months.
2. Molecular analysis for detecting immunoglobulin heavy-chain variable (IGHV) gene mutation status
Molecular analysis assesses the mutation status of the IGHV gene and is an important predictor of survival outcomes and guides prognosis. Unmutated IGHV is associated with a higher risk of adverse genetic mutations, indicative of a poor prognosis, and significantly decreased survival. The mutation status of IGHV genes influences the natural history of CLL with mutated cells exhibiting stability or slower growth compared to unmutated cells. IGHV status remains stable over time and after treatment.
3. Comorbidities and performance status
Because CLL is diagnosed mainly in older adults, comorbidities are commonly present. Multiple comorbidities (two or more) are an independent predictor of clinical outcome, independent of age or disease status. A standardised assessment of performance status, using a tool such as the Eastern Cooperative Oncology Group (ECOG) tool, is recommended to comprehensively evaluate the patient’s overall health and wellbeing. This evaluation is crucial for informing treatment decisions and anticipating potential challenges associated with co-existing health conditions.
Staging
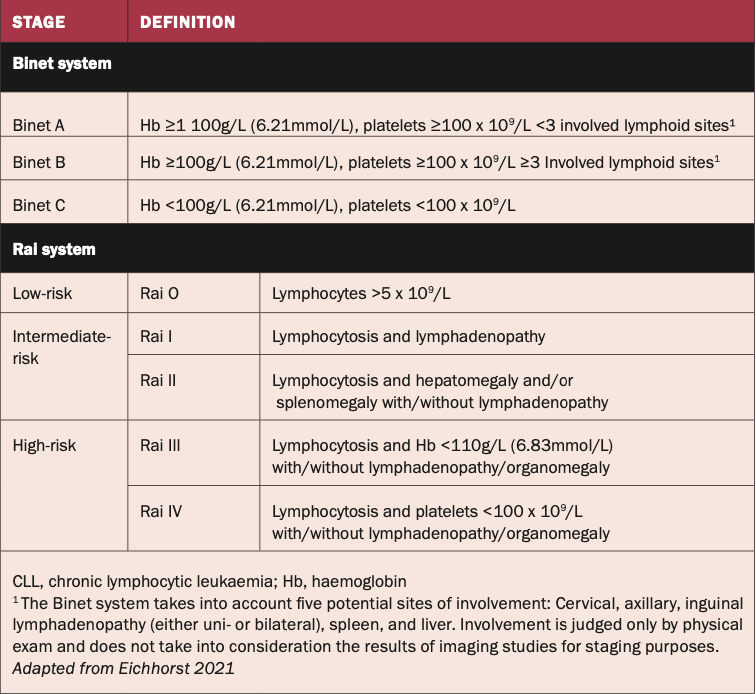
Patients with an established diagnosis of CLL should undergo risk stratification through established staging systems, crucial for both individualised care and clinical trial standardisation. Two widely accepted longstanding clinical staging systems, Rai7 and Binet,8 provide a structured framework based on physical examination and standard laboratory test results. Both systems describe three major subgroups with distinct clinical outcomes.
The Rai system is based on lymphocytosis, hepatomegaly, and splenomegaly with/without lymphadenopathy and haemoglobin and platelet levels. It is more commonly used in the US.
The Binet staging system stratifies patients based on the number of involved lymphoid sites, the level of haemoglobin and platelets, and is more commonly used in the UK and Europe.
Early asymptomatic-stage disease, as determined by either of the staging systems, does not require further risk assessment. See Figure 1.
Figure 1: Staging systems for CLL
Indications for treatment
The criteria for initiating CLL treatment rely on the RAI and Binet staging systems and on the presence of symptoms caused by the disease. Current treatment guidelines from the International Workshop on Chronic Lymphocytic Leukaemia (iwCLL) recommend treatment only for active, symptomatic disease defined by at least one of the criteria outlined in Table 1.5
Table 1: iwCLL treatment guidelines for CLL5
Management options
Despite many advances in the CLL treatment, it remains an incurable disease. The main purpose of systemic therapy is for symptomatic relief and prolonged remission and survival.
CLL treatment decision-making depends a number of factors:6
1. Genetic factors: TP53 mutation and/or deletion and IGHV mutation status significantly shapes treatment strategies.
2. Patient characteristics: Treatment decisions hinge on the patient’s age, overall health, and co-morbidities. These aspects critically influence the feasibility and intensity of therapeutic interventions.
3. Disease stage and symptoms: The disease stage and presence of symptoms contribute to the formulation of an effective treatment plan. Understanding the extent of CLL progression guides the choice between active surveillance and therapeutic intervention.
4. Drug availability and accessibility: The availability of specific drugs and their accessibility also plays a pivotal role in treatment decisions. Treatment plans are tailored based on the pharmacological options accessible in the healthcare environment.
5. Individual patient preference and potential of treatment adherence: Acknowledging individual patient preferences and assessing the potential for treatment adherence is integral to developing a patient-centred approach. Collaboration between healthcare teams and patients is paramount in determining the most suitable treatment plan aligned with the patient’s individual circumstances also.
6. Overall treatment goals: The primary goals of CLL management include achieving symptomatic relief, inducing prolonged remission, and extending overall survival. These goals underscore the necessity of a holistic and personalised approach to treatment.
Watch and wait (active surveillance)
In the case of patients with asymptomatic early-stage disease,7,8 the approach is monitoring without therapy.5 Early intervention therapy is currently not indicated and standard-of-care for these patients is active surveillance aligning with several studies demonstrating no survival benefit in treating patients with early-stage disease. A small subset of patients with CLL will never require treatment and risks associated with unnecessary treatment can be avoided.
During this observation period, patients have a clinical examination, inclusive of measurement of lymph nodes and spleen size, focused history taking to assess disease-related symptoms, and blood tests at three-monthly intervals for the first year. Following these evaluations at the end of a 12-month period, subsequent follow-ups are adjusted to six-to-12-monthly intervals, depending on the burden and dynamics of the disease.
Patient education is integral during this phase, encompassing comprehensive information about the nature of CLL, the indications for treatment, infection risk, and reassurance. This patient-centred approach acknowledges the individualised nature of CLL progression and empowers the patient with a clear understanding of their condition and the rationale behind the surveillance strategy. This proactive plan aims to optimise outcomes while minimising unnecessary medical interventions.
Patients with active disease
The treatment landscape for CLL has rapidly evolved and expanded over the past decade, marked by the integration of targeted therapies including BTK inhibitors, P13K inhibitors, and BCL2 inhibitors. In contrast to the historical utilisation of purine analogues (fludarabine) and alkylating agents (bendamustine, chlorambucil, and cyclophosphamide) in combination with the anti-CD20 antibody, rituximab, as standard-of-care for younger and older patients, respectively, the contemporary approach represents a paradigm shift. The contemporary approach for first-line treatment for CLL uses different combinations of targeted therapies and immunotherapies to achieve better treatment outcomes while minimising side-effects.
Chemotherapy
Historically, chemo-immunotherapy was a common approach for CLL treatment. The first-line treatment regimen of fludarabine, cyclophosphamide, and rituximab (FCR) was considered the standard-of-care for medically-fit patients of <65 years, and bendamustine and rituximab (BR) was the preferred option for older, less fit patients. Newer treatments have largely replaced chemo-immunotherapy due to their enhanced safety and efficacy profiles.
Targeted therapies
BTK inhibitors including ibrutinib, acalabrutinib, and zanubrutinib, represent a class of oral drugs which block the activity of Bruton’s tyrosine kinase (BTK), a protein necessary for the survival of CLL cells. BTK inhibitors are used in both elderly and younger populations. They have shown significant effectiveness as continuous monotherapy or in combination with anti-CD20 antibody therapies or BCL-2 inhibitors. They are indicated as first-line therapy in CLL with TP53 mutation or 17p deletion where BTK inhibitors have shown remarkable clinical effectiveness. The advent and integration of BTK inhibitors into CLL treatment protocols were a significant advancement, offering clinicians a targeted and potent therapeutic option to disrupt the survival pathways of CLL cells.
BCL-2 inhibitors including venetoclax represent a class of oral anti-cancer drugs that inhibit the BCL-2 protein, which prolongs CLL survival. Venetoclax is used as monotherapy and in combination with anti-CD20 antibody therapy or BTK inhibitor Ibrutinib, often prescribed as a fixed duration therapy regimen. The precision of venetoclax in inhibiting the BCL-2 protein represents a further therapeutic advancement disrupting the survival mechanisms of CLL cells.
PI3K inhibitors are oral drugs, including idelalisib and duvelisib, which target the phosphoinositide 3-kinase (PI3K) pathway, a critical regulator of cell growth and survival. They are primarily used in the management of relapsed or refractory CLL.
Immunotherapy
Monoclonal antibodies (rituximab, binutuzumab): Monoclonal antibodies including rituximab and obinutuzumab, represent a targeted immune therapeutic approach by specifically engaging CD20 proteins, found on the surface of CLL cells. This targeted recognition facilitates the immune system in identifying and eliminating CLL cells. They are used in combination with targeted therapies and chemotherapy.
CAR T-cell therapy: Chimeric antigen receptor (CAR) T-cell therapy is an innovative approach involving genetic modification of a patient’s T-cells to selectively target and eliminate CLL cells. This advanced therapy, which is available in Ireland, has shown efficacy in a number of haematological malignancies, but remains investigational for CLL.9
Stem cell transplant: In certain cases, particularly among younger patients with aggressive disease, a stem cell transplant may be considered. This is a high-risk procedure and is typically reserved for cases that have demonstrated sub-optimal response to other treatments or in cases of Richter’s transformation (the development of an aggressive large-cell lymphoma in the setting of underlying CLL).
Treatment of relapsed disease
Disease recurrence, even after successful treatment, is a reality for many CLL patients. Consistent with first-line therapy principles, the initiation of subsequent treatment is reserved for symptomatic patients rather than at first reappearance of disease.8 In the case of rapid progression while on targeted agents, an immediate change of therapy is recommended.
The selection of relapse treatment is a nuanced process, guided by several key considerations. These include the intensity of, and side-effects of, previous therapies, the duration of the previous response, the risk of complications, current evidence, and patient preference. Genetic defects should also be reassessed by FISH to identify if any genetic evolution has occurred such as the acquired 17P deletion.
Participation in clinical trials is a valuable option for patients with CLL. It offers access to targeted treatment options and combinations being investigated for efficacy and safety profiles and the comparison of fixed duration versus continuous therapy in CLL. Most recently the European CLL 17 trial was opened to Irish patients.11 This randomised prospective trial is investigating the outcome of ibrutinib monotherapy versus fixed duration venetoclax plus obinutuzumab versus fixed duration ibrutinib plus venetoclax in previously untreated CLL patients. By actively engaging in such clinical trials, patients not only contribute to the collective understanding of CLL management, but also gain access to potentially new intervention strategies for CLL.
Disease-related complications of CLL
Infection risk
People with CLL have both disease-related and treatment-related immunosuppression and are at risk of infections, often severe and sometimes fatal.
Vaccinations
Annual influenza vaccination, PPV23 pneumococcal vaccination, and Covid-19 vaccination are advised as per HSE immunisation schedules. Live vaccines (measles/mumps/rubella, live polio, yellow fever, and varicella vaccine [Zostavax]) should not be given. Patients should avoid contact with children who have received the live nasal influenza vaccine for seven days. The recombinant varicella vaccine (Shingrix) is safe for patients with CLL. Patients should be advised that their immune response to vaccinations is lower than the general population and they should avoid exposure to infections.
Anti-infective prophylaxis: Anti-bacterial prophylaxis should be considered for all patients with a history of recurrent or serious bacterial infections. In patients receiving therapies, prophylaxis against P jirovecii, and viral infections is also considered.
Immunoglobulin replacement therapy: Hypogammaglobinaemia is a common finding in patients with CLL. There is also the risk of iatrogenic secondary immunodeficiency from treatments for CLL.
Immunoglobulin (IgG) replacement therapy is indicated for patients who experience recurrent or severe bacterial infections despite six months of continuous oral antibiotic therapy and have a total IgG <4g/l.12 IgG is a pharmaceutical product derived from pooled plasma. A starting dose of 0.4-0.6g/kg/month is recommended with adjustment according to the trough IgG level. Current licenced products available in Ireland include intravenous preparations, subcutaneous preparations, and hyaluronidase-facilitated products, which have similar efficacy in terms of infection prevention.
Covid-19 (Coronavirus): The Covid-19 pandemic presented particular challenges for patients with CLL. It is likely that the secondary immunodeficiency associated with CLL confers a higher risk of severe Covid-19 disease requiring hospitalisation. The degree of protection afforded to patients with CLL by the available Covid-19 vaccines is lower than that of healthy age-matched controls. Vaccination is recommended to all patients with CLL, ideally completed before commencing therapy. Treatment for Covid-19 is currently indicated for patients with chronic B-cell lymphoproliferative disorders and those who have received B-cell depleting therapy, including anti-CD20 therapy, in the previous 12 months. Treatment options include intravenous sotrovimab, remdesivir, and oral Paxlovid.
Risk of secondary primary cancers
People with CLL have an increased risk of developing secondary primary cancers.13 Age-adapted screening of common cancers including breast, bowel, and cervical cancer is recommended to facilitate early detection and treatment. Skin cancer prevention and self-monitoring advice should also be given.
Follow-up and survivorship for CLL
Continued monitoring through life-long observation is recommended for all people diagnosed with CLL. It is important to educate patients about the prescribed follow-up schedule and underscore the importance of consistent follow-up care. During these follow-up visits, a comprehensive approach is taken, involving a focused medical history, FBC, and examination of lymph nodes, liver, and spleen. The frequency of these assessments typically ranges from every three-to-12 months, tailored to the specific dynamics of the disease and prior treatment.
In addition to routine medical assessments, follow-up care extends to addressing long-term issues related to CLL and its treatment. Recognising the chronic nature of CLL, it is crucial to acknowledge the impact on patients’ quality-of-life. Efforts should be made to support patients’ psycho-social wellbeing. This can be achieved by referring individuals to reliable information sources, CLL patient support and advocacy groups, and, when appropriate, psycho-social health teams. This holistic approach aims to provide comprehensive care beyond the immediate medical aspects, recognising the broader challenges and considerations associated with living with a chronic illness such as CLL.
References
National Cancer Registry Ireland. Cancer in Ireland 1994-2018 with estimates for 2018-2020: Annual report of the National Cancer Registry. NCRI, Cork, Ireland. [Internet]. 2020. Available at: https://ncri.ie/data
Waldron C, O’Brien D, Brophy S, et al. Epidemiology of chronic lymphocytic leukaemia in an Irish subpopulation with total case ascertainment: An additional tool for health economic planning. Br J Haematol. 2022; 196(5):e47-e49.
SEER*Explorer: An interactive website for SEER cancer statistics [Internet]. Surveillance Research Programme, National Cancer Institute; 2023 Apr 19. [updated: 2023 Jun 8; cited 2023 Aug 21]. Available at: https://seer.cancer.gov/statistics-network/explorer/
Cerhan JR, Slager SL. Familial predisposition and genetic risk factors for lymphoma. Blood. 2015; 126(20):2265-2273.
Hallek M, Cheson BD, Catovsky D, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018; 131(25):2745-2760
Wierda WG, Brown J, Abramson JS, et al. NCCN guidelines insights: Chronic lymphocytic leukaemia/small lymphocytic Lymphoma, version 3.2022. J Natl Compr Canc Netw. 2022; 20(6):622-634.
Rai KR, Sawitsky A, Cronkite EP, et al. Clinical staging of chronic lymphocytic leukaemia. Blood. 1975;46:219-234
Binet JL, Auquier A, Dighiero G, et al. A new prognostic classification of chronic lymphocytic leukaemia derived from a multivariate survival analysis. Cancer. 1981; 48:198-206
Shadman M. Diagnosis and treatment of chronic lymphocytic leukaemia: A review. JAMA. 2023; 329(11):918-932 10. HSE National Cancer Control Programme. National SACT regimens. Available at: www.hse.ie/eng/services/list/5/cancer/profinfo/chemoprotocols/leukemia-bmt/protocols%20leukaemia.html 11. Cancer Trials Ireland DSSG Group: Chronic lymphocytic leukaemia (CLL) CLL 17: Previously available at: www.cancertrials.ie/current-trials/lymphoma-blood-cancers/chronic-lymphocytic-leukaemia-cll/ 12. HSE National Cancer Control Programme. Guidance Document: Patient selection for the use of immunoglobulin replacement therapy in cancer patients with secondary immunodeficiency. Available at: www.hse.ie/eng/services/list/5/cancer/profinfo/medonc/sactguidance/nccp-guidance-final-patient-selection-for-immunoglobulin-replacement-therapy.pdf 13. Eichhorst B, Robak T, Montserrat E, et al. Chronic lymphocytic leukaemia: ESMO clinical practice guidelines for diagnosis, treatment, and follow-up. Ann Oncol. 2021; 32(1):23-33












Leave a Reply
You must be logged in to post a comment.